Ciencia y Salud, Vol. 10, No. 1, enero-abril, 2026 • ISSN (impreso): 2613-8816 • ISSN (en línea): 2613-8824
LEUCEMIA PROMIELOCÍTICA AGUDA CON COMPROMISO AL SISTEMA NERVIOSO CENTRAL: A PROPÓSITO DE UN CASO CLÍNICO
Acute promyelocytic leukemia with central nervous system involvement: a clinical case report
DOI: https://doi.org/10.22206/cisa.2026.v10i1.3238
Leonel De Jesús Camacho Castillo1
Recibido: 16/07/2024 • Aceptado: 08/06/2025

Cómo citar: Camacho Castillo, L. J. (2026). Leucemia promielocítica aguda con compromiso al sistema nervioso central: a propósito de un caso clínico. Ciencia y Salud, 10(1), 77-92. https://doi.org/10.22206/cisa.2026.v10i1.3238
Resumen
Introducción: La leucemia es una neoplasia hematológica que engloba diversos subtipos siendo la Leucemia Promielocítica Aguda (LPA) una de las más agresivas, caracterizada por la afectación del linaje mieloide. Esta condición presenta una alta mortalidad si no es diagnosticada y tratada a tiempo. Aunque a LPA clásicamente se presenta con manifestaciones hemorrágicas y alteraciones hematológicas, existen casos con presentaciones clínicas inusuales, incluyendo manifestaciones neurológicas atípicas. Objetivo: Presentar el caso de una paciente con diagnóstico de LPA; destacar la importancia de un diagnóstico temprano y un manejo adecuado, así como enfatizar las complicaciones graves asociadas con la enfermedad y su tratamiento. Métodos y materiales: Se realizó un análisis exhaustivo de los estudios clínicos y paraclínicos, incluyendo hemograma, frotis de sangre periférica, aspirado de médula ósea, pruebas de inmunofenotipificación y diagnóstico molecular mediante FISH. El tratamiento inicial consistió en la administración de ácido holo-transretinoico (ATRA) y quimioterapia dirigida. Resultados: La paciente, una femenina de 36 años, con antecedentes de síndrome de ovarios poliquísticos, menorragia, anemia recurrente y toxicomanía por cocaína, presentó áreas de equimosis difusas, paraparesia inferior y alteraciones hematológicas significativas. A pesar de la instauración temprana de tratamiento en un centro especializado, falleció debido a complicaciones derivadas de la LPA y el síndrome de diferenciación asociado al tratamiento. Conclusión: Este caso enfatiza la trascendencia de un diagnóstico precoz y un manejo multidisciplinario de la LPA, especialmente en su presentación atípica. La rápida progresión de la enfermedad y las complicaciones derivadas del tratamiento subrayan la necesidad de un enfoque integral y oportuno para mejorar los resultados clínicos en pacientes con esta neoplasia.
Palabras clave: Leucemia promielocítica aguda, médula ósea, sistema nervioso central, neoplasias hematológicas, anemia.
Abstract
Introduction: Leukemia is a hematological neoplasm that encompasses various subtypes, with Acute Promyelocytic Leukemia (APL) being one of the most aggressive, characterized by the involvement of the myeloid lineage. This condition has a high mortality rate if not diagnosed and treated in a timely manner. Although APL classically presents with hemorrhagic manifestations and hematological alterations, there are cases with unusual clinical presentations, including atypical neurological manifestations. Objective: To present the case of a patient diagnosed with APL; to highlight the importance of early diagnosis and proper management, as well as to emphasize the serious complications associated with the disease and its treatment. Methods and materials: A comprehensive analysis of clinical and paraclinical studies was performed, including complete blood count, peripheral blood smear, bone marrow aspirate, immunophenotyping tests, and molecular diagnosis by FISH. Initial treatment consisted of the administration of all-trans retinoic acid (ATRA) and targeted chemotherapy. Results: The patient, a 36-year-old female with a history of polycystic ovary syndrome, menorrhagia, recurrent anemia, and cocaine addiction, presented with areas of diffuse ecchymosis, inferior paraparesis, and significant hematologic abnormalities. Despite early treatment at a specialized center, she died due to complications associated with APL and treatment-associated differentiation syndrome. Conclusion: This case emphasizes the importance of early diagnosis and multidisciplinary management of APL, especially in its atypical presentation. The rapid progression of the disease and treatment-related complications underscore the need for a comprehensive and timely approach to improve clinical outcomes in patients with this neoplasm.
Keywords: Acute promyelocytic leukemia, bone marrow, central nervous system, hematologic malignancies, anemia.
Introducción
Las leucemias se definen como un grupo de alteraciones oncológicas heterogéneas con características morfológicas, clínicas, citogenéticas y pronósticos diversos de la médula ósea y de las células de la sangre. Según su tipo, se clasifican en: leucemia mieloide aguda, leucemia mieloide crónica, leucemia linfoblástica aguda y leucemia linfocítica crónica, cada una con subtipos específicos1.
La leucemia promielocítica aguda corresponde al subtipo M3 de la leucemia mieloide aguda según la clasificación de la Asociación Franco-Américo-Británica (FAB), la cual se origina por una traslocación balanceada entre los cromosomas 15 y 17; involucra al gen que codifica para el receptor alfa del ácido retinoico (RARA) en el cromosoma 17 y el de la leucemia promielocítica en el cromosoma 15, lo que da origen a la traslocación t(15;17) PML/RARA2.
Esta entidad se describió por primera vez en 1957 en pacientes con tendencia hemorrágica grave, fibrinólisis, rápido deterioro del estado clínico y presencia de promielocitos en sangre periférica y médula ósea. Es relativamente rara y comprende alrededor del 7% al 8% de los casos de leucemia mieloide aguda en adultos. Suele observarse en personas de mediana edad con una edad media de 47 años, ocurriendo muy raramente antes de los 20 años. Su incidencia es ligeramente mayor en hombres que en mujeres3.
Los pacientes suelen presentar debilidad y fatiga generalizadas, sangrado gingival, petequias o equimosis, cambios visuales secundarios a hemorragias retinianas, epistaxis o menorragia o infecciones. También complicaciones trombóticas como trombosis venosa profunda, embolia pulmonar, accidente cerebrovascular, coagulación intravascular diseminada manifiesta y sangrado franco. La pancitopenia es común en el momento de la presentación3.
El diagnóstico requiere de un estudio completo de sangre periférica y de médula ósea (punción - biopsia) y deberá incluir análisis morfológico, inmunofenotipo, citogenético-cariotipo y de fluorescencia por hibridación in situ (FISH) y/o anticuerpos monoclonales anti - LPM1.
La leucemia promielocítica aguda (LPA) en ausencia de tratamiento es rápidamente fatal e incluso después del inicio de la terapia. Estando la principal causa de mortalidad temprana relacionada con las complicaciones hemorrágicas4.
La mortalidad temprana en LPA es definida como la muerte dentro de los primeros 30 días de tratamiento y representa el principal problema de esta entidad. Es causada principalmente por una hemorragia en relación a la coagulación intravascular diseminada (CID), fibrinólisis y plaquetopenia severa; además de otras causas como el síndrome de diferenciación y las infecciones. Diversos estudios han descrito que los factores de riesgo relacionados a la mortalidad temprana de LPA son la edad mayor a 60 años, sexo masculino, niveles altos de recuento de leucocitos, alto porcentaje de promielocitos en sangre periférica, bajo conteo de plaquetas, función renal alterada y bajos niveles de fibrinógeno al diagnóstico5.
Diferentes investigaciones realizadas con grandes poblaciones han reportado que la tasa de mortalidad temprana en LPA puede variar entre el 5-50%, es decir, aproximadamente 1 de cada 3 pacientes diagnosticados con LPA muere durante el primer mes. Sin embargo, las características y etiología de la mortalidad varían de acuerdo con el tiempo transcurrido desde el inicio inducción; en fases precoces se postula que la mortalidad está relacionada al sangrado, mientras que en fases tardías con infecciones. Es por esto por lo que se ha implementado una tendencia mundial a la reducción de las tasas de mortalidad temprana de LPA mediante la instauración de programas de reducción de riesgos, iniciativas colaborativas para la estandarización del tratamiento y mejoras en el manejo de soporte, así como la administración temprana de ATRA5.
El tratamiento específico incluye el ácido transretinoico (ATRA) que induce la diferenciación granulocítica o el trióxido de arsénico (ATO), el cual recientemente ha mejorado las tasas de curación con menor toxicidad y mortalidad frente a las combinaciones con quimioterapia5. Estudios preclínicos demostraron sinergismo significativo entre ATRA y ATO a través de la inducción de diferenciación celular y apoptosis en leucemia promielocítica6, 7. Además, varios ensayos clínicos de fase temprana informaron resultados favorables usando esta terapia6, 8, 9.
El estudio APL15 demostró que la combinación de ATRA y ATO sin quimioterapia podría ser un tratamiento eficaz y bien tolerado para pacientes con LPA con cualquier factor de riesgo. En esta investigación, la estrategia citorreductora empleada se basó en hidroxiurea, cuya dosis se tituló hasta una dosis máxima de 0,1 g/kg/día. Además, el estudio incorporó el uso de manitol para aumentar la permeabilidad del arsénico en el líquido cefalorraquídeo, con el objetivo de reducir el riesgo de recurrencia en el SNC10, 11.
Presentación de caso clínico
Se trata de paciente femenina de 36 años, G5 P0 C0 A5, de ocupación manicurista, con antecedentes mórbidos conocidos de síndrome de ovarios poliquístico diagnosticado hace 15 años, menorragia y anemia recurrente de igual data con tratamiento no especificado; toxicomanía por cocaína de patrón de uso no esclarecido negando policonsumo; refirió familiar (hermana) y paciente que esta se encontraba en aparente control de sus comorbilidades hasta 7 días previos a su ingreso que inició cuadro clínico caracterizado por episodios febriles, intermitentes, termometrados con valores entre en 38.5 – 39°C, sin predominio de horarios, con escalofríos, diaforéticos, medicados con capletas de antipirético central tipo acetaminofén 500 mg + salicilato tipo ácido acetilsalicílico 81 mg, los cuales cedían para luego volver a reaparecer de igual intensidad; aunado a cefalea holocraneana, de aparición progresiva, de carácter tipo opresivo, fuerte intensidad 10/10 en escala de EVA; acompañado de áreas de equimosis en tronco anterior, posterior y en las extremidades, de tamaños variables, con bordes asimétricos, no concéntricas, no sobreelevadas, motivos por los cuales acudió vía emergencia a un centro de Salud privado en donde permaneció ingresada por 3 días y por razones económicas para la realización de paraclínicas decidió acudir por sus propios medios a la consulta del Departamento de Hematología de Hospital Regional Universitario San Vicente de Paúl con fines de investigación; sin embargo, posterior a la valoración todo cuadro se exacerbó con la aparición de nuevas áreas de equimosis en los antebrazos y cuadrante inferior de mama izquierda y astenia e hiporexia, lo que le motivó acudir vía emergencia de nuestro centro hospitalario donde tras previa evaluación clínica y paraclínica se decidió su ingreso con fines diagnósticos y terapéuticos.
Examen físico:
- Estado general: paciente alerta, orientada en las 3 esferas sensoriales, con buena prosodia del lenguaje, biotipo constitucional mesomorfo, afebril al momento de la evaluación, eupneica, quejumbrosa, leve palidez de piel y mucosas. Con los siguientes signos vitales: TA 120/70 mmHg, FC 90 lpm, FR 20 rpm, SpO2 94%, Temp 37.1°C.
- Cabeza: normocéfala, pelo de buena implantación y distribución, sin reblandecimiento óseo, sin hundimiento ni protrusiones.
- Ojos: simétricos, pupilas isocóricas, fotorreactivas, reflejo corneal presente, escleras anictéricas, conjuntivas pálidas, no edema palpebral ni secreciones.
- Nariz: tabique nasal central, no aleteo nasal, narinas permeables, vibrisas presentes, no pólipos ni salida de secreción.
- Boca: labios simétricos, mucosa oral húmeda, edéntula parcial, caries dentales difusas, amígdalas eutróficas, lengua normoglosa, paladar duro y blando sin lesiones, úvula central.
- Cuello: cilíndrico, móvil, tráquea central, pulsos carotideos bilaterales de buena amplitud y forma, no soplos, no adenopatías palpables, no ingurgitación yugular.
- Tórax: simétrico, normodinámico, con áreas de equimosis en tronco anterior y posterior y cuadrante inferior de mama izquierda, de tamaños variables, de bordes asimétricos, no concéntricas, no sobreelevadas; no retracciones intercostales ni subcostales, ápex cardíaco en 5to espacio intercostal línea medio clavicular izquierda, frémito táctil bilateral conservado.
- Corazón: Ruidos cardiacos regulares, R1-R2 íntegros, no galope auricular ni ventricular. Soplo mesosistólico en foco mitral 3/6 en Escala de Levine.
- Pulmones: normoventilados, murmullo vesicular audible, no estertores agregados.
- Abdomen: globoso a expensas de panículo adiposo, cicatriz en hipocondrio derecho por colecistectomía previa, peristalsis 3/5, matidez hepática conservada, timpanismo en resto del abdomen, depresible, no doloroso a la palpación superficial ni profunda, no masas, no visceromegalias palpables.
- Piel y anexos: fototipo III en escala de Fitzpatrick, turgor y elasticidad acorde a la edad, presencia de áreas de equimosis difusas en tronco y extremidades.
- Genitales externos: fenotípicamente adecuados para sexo y edad.
- Extremidades superiores: simétricas, con áreas de equimosis de tamaños variables, de bordes asimétricos, no concéntricas, no sobreelevadas, móviles, pulsos periféricos palpables en buena forma y amplitud, no edema.
- Extremidades inferiores: simétricas, con áreas de equimosis de tamaños variables, de bordes asimétricos, no concéntricas, no sobreelevadas, móviles, pulsos periféricos presentes en buena forma y amplitud, no edema.
- Neurológico: alerta, con nivel de conciencia conservado. Orientada en tiempo, espacio y persona. Lenguaje fluido, con prosodia, articulación y comprensión preservadas; sin disartria ni afasia aparentes. Memoria anterógrada y retrógrada integra. Juicio crítico, analítico y capacidad de abstracción conservados. Atención y concentración sin alteraciones evidentes. No signos de irritación meníngea: rigidez de nuca, signos de Kernig y Brudzinski negativos. Pares craneales del I al XII sin alteraciones clínicamente evidentes. Campimetría por confrontación sin defectos aparentes; pupilas isocóricas y normorreactivas; movimientos oculares extrínsecos completos, sin nistagmo ni diplopía; reflejo corneano presente y simétrico; fuerza de elevación del paladar y movilidad lingual simétrica. Sensibilidad superficial (tacto, dolor y temperatura) y profunda (vibratoria, propioceptiva) conservadas en las cuatro extremidades. Fuerza muscular 5/5 en Escala de Daniels en todas las extremidades de forma global y simétrica. No se observó fatigabilidad ni paresias. Reflejos osteotendinosos presentes, simétricos y normoactivos: bicipital ++, tricipital ++, estilorradial ++, rotuliano ++, aquíleo ++ bilateralmente. Clonus ausente. Reflejos cutaneoabdominales presentes y simétricos. Coordinación adecuada. Pruebas cerebelosas (dedo-nariz, talón-rodilla, prueba de diadococinesia) sin dismetría ni disdiadocinesia. Marcha estable, con adecuado balance y sin ataxia. No se observaron signos de lateralización neurológica.
En concordancia con el examen fisico previamente descrito, la Figura 1 documenta los hallazgos clínicos de la paciente al momento de su llegada al servicio de emergencia.
Figura 1. Momento de llegada de la paciente al servicio de emergencia
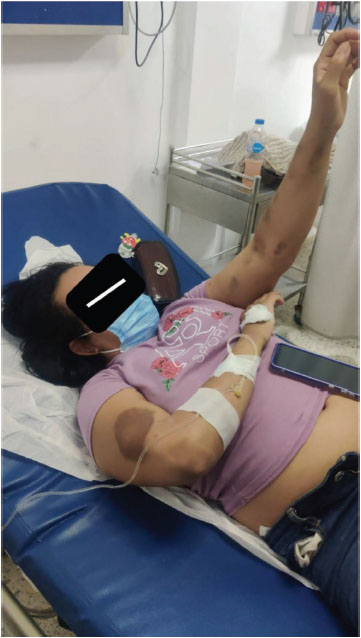
Fuente: Imagen original capturada por el autor. Publicación realizada con autorización expresa de la paciente y familiares, en conformidad con principios éticos de confidencialidad y consentimiento informado.
Durante su estancia hospitalaria, se realizó una documentación fotográfica complementaria con fines de seguimiento y evaluación de las lesiones (Figura 2), como parte del monitoreo clínico continuo.
Figura 2. Secuencia fotográfica durante la estancia de la paciente en sala clínica
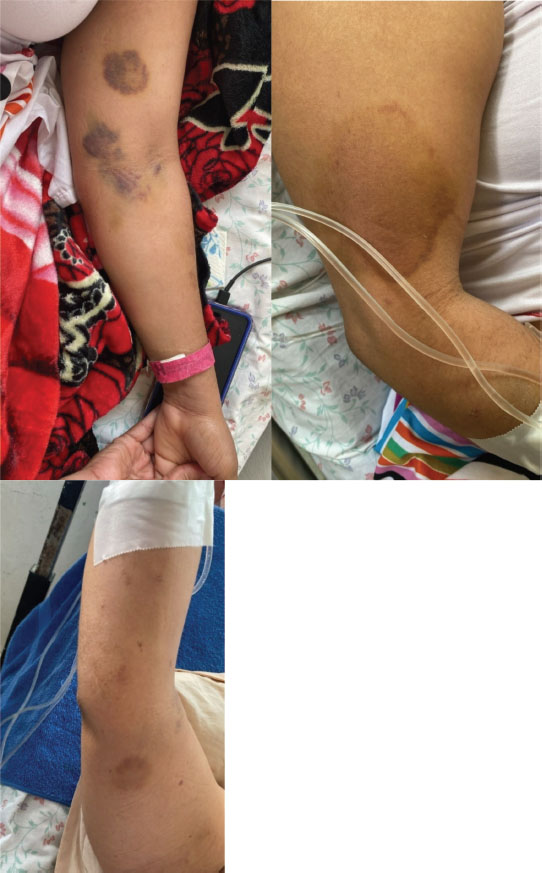
Fuente: Imágenes originales capturadas por el autor. Publicación realizada con autorización expresa de la paciente y familiares, en conformidad con principios éticos de confidencialidad y consentimiento informado.
Como parte de la evaluación diganóstica durante su hospitalización, se realizaron estudios de laboratorio y de imagen (Tabla 1), los cuales orientaron a su abordaje posterior.
Tabla 1. Resultados de exámenes paraclínicos y estudios imagenológicos durante la hospitalización
Hemograma: WBC 82.32 10^3/ul, RBC 3.38 10^6/ul, HGB 9.5 g/dl, HCT 28.9%, MCV 65.5 fl, MCH 28.1 pg, MCHC 32.9 g/ul, PLT 64 10^3/ul, MPV 10.3 fl, LYMPH% 10.7%, MON% 67.9%, NEU% 0.9%, EOS% 0.0%, BAS% 0.1%, LYMPH# 8.82 10^3/ul, MON# 72.36 10^3/ul, NEU# 0.70 10^3/ul, EOS 0.00 10^3/ul, BAS 0.10 10^3/ul. |
Tipificación sanguínea: B RH positivo. |
Químicas: glucosa 130 mg/dl, colesterol 215 mg/dl, triglicéridos 316 mg/dl, C-HDL 48.1 mg/dl, C-LDL 103.7 mg/dl, VLDL 63.2 mg/dl, ácido úrico 6.42 mg/dl, creatinina 0.92 mg/dl, urea 24.4 mg/dl, BUN 11.4 mg/dl, albumina 4.4 g/dl, bilirrubina total 0.27 mg/dl, bilirrubina directa 0.07 mg/dl, bilirrubina indirecta 0.20 mg/dl, proteínas totales 0.27 mg/dl, globulina 2.5 g/dl, relación A/G 1.8 g/dl, TGO 75 u/l, TGP 109 u/l, amilasa 58 u/l, lipasa 27.0 u/l, TP 13.7 seg, TPT 22.8 seg, T3 total 0.80 nmol/l, T4 total 6.2 ug/dl, TSH 1.49 ui/ml, ferritina 105.4 ng/ml, vitamina B12 69.00 pg/ml, virales negativos, sodio 140 mmol/l, potasio 3.33 mmol/l, calcio 8.41 mg/dl, cloro 98 mmol/l. |
Gases arteriales: pH 7.48, pO2 55 mmHg, pCO2 20.7 mmHg, CH+, r 33.0 mmol/L, HCO3-act, r 15.1 mmol/L, HCO3-std, r 19.8 mmol/L, BE (ecf), r -8.4 mmol/L, BE (B), r -5.6 mmol/L, BB (B), r 42.9 mmol/L, pO2 (A-a), r 70 mmHg, pO2 (a/A), r 0.44 mmHg. |
Examen de orina: color amarillo, olor suis generis, aspecto ligero turbio, pH 7.0, densidad 1.010, proteinuria +, cetonuria +, bilirrubina +, leucocitos 0-2/c, hematíes 0-2/c, células epiteliales algunas, bacterias algunas. |
Electrocardiograma: con ritmo sinusal. |
Ecocardiograma: válvula mitral engrosada con insuficiencia leve. Aorta 30 mm, pared posterior 10 mm, septum interventricular 10 mm, aurícula izquierda 14 cm2, aurícula derecha 13 cm2, ventrículo izquierdo 38-22 mm, ventrículo derecho 28 mm, fracción de acortamiento 41%, fracción de eyección 75%, TAPSE 28 mm. |
Doppler carotídeo: sin hallazgos patológicos. |
Radiografía de tórax: con presencia de trama para-hiliar derecha engrosada. |
Tomografía de cráneo: con área hipodensa, circunscrita, de bordes simétricos, en territorio de M1- M2 derecha (Figura 4). |
Fuente: Construcción propia a partir del expediente clínico. Datos utilizados con autorización expresa de la paciente y sus familiares, conforme a los principios éticos de confidencialidad y consentimiento informado.
Ante los hallazgos clínicos y paraclínicos, decidimos realizar un frotis de sangre periférica para evaluar la presencia de posibles células anormales. En el análisis, se identificaron blastos (Figura 3), lo que sugirió un trastorno hematológico que requería una evaluación diagnóstica adicional para determinar su naturaleza y proceder con tratamiento adecuado.
Figura 3. Frotis de sangre periférica con presencia de blastos
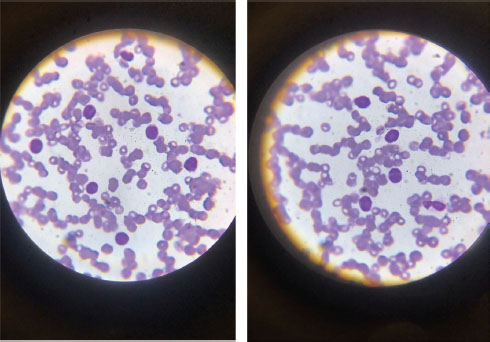
Fuente: Imágenes originales obtenidas por el autor durante la evaluación hematológica. Publicación realizada con autorización de la paciente y familiares, conforme a los principios éticos de confidencialidad y consentimiento informado.
Al cuarto día de su ingreso hospitalario, la paciente presentó un cuadro caracterizado por paraparesia inferior de aparición insidiosa, además de la sintomatología por lo que fue ingresada inicialmente. Esto nos planteó la posibilidad de una lesión neurológica focalizada en la médula espinal o estructuras supramedulares; ante la evolución del cuadro y la aparición del déficit motor, se realizó una tomografía axial computarizada de cráneo sin contraste, cuyos hallazgos (Figura 4) mostraron un área de hipodensidad en los territorios M1 y M2 correspondientes a la arteria cerebral media derecha, lo que objetivó la presencia de una lesión isquémica en dicha distribución, clinicamente concordante con el compromiso motor evidenciado.
Figura 4. Tomografía axial de cráneo sin contraste con área de hipodensidad en los territorios M1 – M2 de la arteria cerebral media derecha
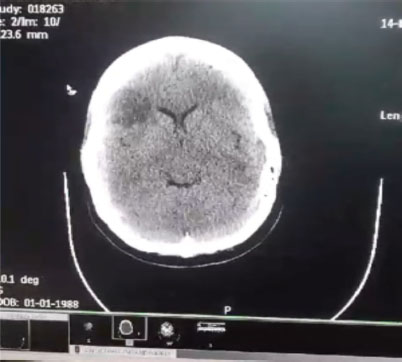
Fuente: Tomografía institucional registrada durante la hospitalización. Uso aprobado por la paciente y familiares conforme a normas éticas de confidencialidad.
Durante su internamiento en nuestro hospital, la paciente fue tratada por el departamento de Hematología con: solución salina 0.9% 3000 ml cada 24 horas vía endovenosa, omeprazol 40 mg cada 24 horas vía endovenosa, paracetamol 1 gramo cada 8 horas vía endovenosa, bicarbonato de sodio 1 ampolla cada 24 horas vía endovenosa, alopurinol 300 mg cada 24 horas vía oral, dexametasona 4 mg cada 12 horas vía endovenosa, ceftriaxona 1 gramo cada 12 horas vía endovenosa. Se Transfundió una unidad de paquete de plaquetas por aféresis y un paquete globular isogrupo y tipo, de sangre fresca, tras previo cruce sanguíneo y premedicación.
En el contexto de las manifestaciones neurológicas y tras la identificación de una lesión hipodensa en los territorios M1-M2 de la arteria cerebral derecha en la tomografía axial de cráneo sin contraste; se solicitó interconsulta al servicio de Neurología para valoración especializada. Luego de la evaluación clínica y revisión de los estudios por imagen, el equipo neurológico consideró que los hallazgos eran compatibles con una enfermedad vasculocerebral de tipo isquémico en fase aguda versus subaguda, por lo que se instauró tratamiento dirigido a la misma, incluyendo medidas de soporte, control de factores de riesgo vascular y prevención secundaria.
En paralelo, se solicitó interconsulta al servicio de Psicología Clínica con el propósito de brindar atención especializada en salud mental, dado el contexto de enfermedad potencialmente grave y la carga emocional asociada al proceso diagnóstico y terapéutico. Se consideró necesario ofrecer intervención psicológica oportuna, orientada a proporcionar contención emocional, favorecer mecanismo de afrontamiento adaptativos y prevenir el desarrollo de síntomas afectivos como ansiedad, depresión o desregulación emocional. La evaluación también tuvo como finalidad explorar el impacto subjetivo de la hospitalización, las percepciones del diagnóstico por parte de la paciente, su red de apoyo psicosocial y su disposición para la adherencia al tratamiento, con vistas a implementar un plan de intervención acorde a sus necesidades emocionales durante la hospitalización y en el seguimiento subsecuente.
Posteriormente, y con el objetivo de profundizar en la caracterización del cuadro hematológico, se llevó a cabo un aspirado de médula ósea como parte del abordaje diagnóstico especializado. Este procedimiento se indicó ante la persistencia de alteraciones hematimétricas significativas y la presencia de blastos en sangre periférica, lo que motivó la necesidad de evaluar directamente la celularidad y arquitectura medular. La muestra obtenida fue remitida para estudio morfológico, citológico, y eventualmente, inmunofenotipo y citogenético, a fin de establecer con precisión el tipo de estirpe celular comprometida, su grado de maduración y las posibles alteraciones compatibles.
Los hallazgos reportados en el estudio del aspirado resultaron fundamentales para consolidar la orientación diagnóstica, permitiendo correlacionar los datos clínicos, paraclínicos y microscópicos en la definición del proceso patológico subyacente y en la toma de decisiones terapéuticas; para este propósito, se integraron los resultados del análisis por citometría de flujo para leucemia aguda (Tabla 2), junto con los estudios de oncopatología molecular, incluyendo el FISH PML/RAR (Tabla 3) y el FISH para ETO/AML1 (Tabla 4), los cuales se presentan a continuación.
Tabla 2. Análisis por Citometría de Flujo para Leucemia aguda
Anticuerpo |
Descripción |
Resultados |
CD2 |
Antígeno Pan-T, células NK |
Positivo/Dim |
CD3 |
Antígeno Pan-T, complejo TCR |
Negativo |
CD4 |
Subconjunto T- Helper, correceptor MHC-II, expresión débil monocitos |
Negativo |
CD5 |
Marcador de células T, subtipo de linfoma de células B |
Negativo |
CD7 |
Antígeno Pan-T, células NK, precursores mieloides tempranas |
Negativo |
CD8 |
Linfocitos T supresores/citotóxicos, células litorales esplénicas |
Negativo |
CD56 |
Células NK |
Negativo |
CD57 |
Marcador neuroendocrine, células NK |
Negativo |
CD45 |
Antígeno panleucocitario |
Positivo/moderado |
CD10 |
Precursor linfomas/leucemias, Hematogonas, granulocitos maduros |
Negativo |
CD19 |
Antígeno pan-B; débil o negativo en células plasmáticas |
Negativo |
CD20 |
Células B maduras |
Negativo |
CD23 |
Marcador de células B |
Negativo |
CD38 |
Células progenitoras linfoides, blastos, células plasmáticas |
Positivo/Dim |
sKappa |
Linfocitos B |
Negativo |
sLambda |
Linfocitos B |
Negativo |
CD11b |
Monocitos, células mieloides, células NK, subgrupo de linfocitos T y B |
Negative |
CD13 |
Células mieloides tempranas y maduras y monocitos, subgrupo de células LGL |
Positivo/Dim |
CD14 |
Monocitos: Fuerte; células mieloides: débil |
Negativo |
CD16 |
Células mieloides maduras, células NK |
Negativo |
CD33 |
células mieloides, disminución de la intensidad con la maduración |
Positivo/Dim |
CD34 |
Células madre, blastos |
Positivo/Dim |
CD64 |
Monocitos y células mieloides de linaje temprano |
Negativo |
CD117 |
c-kit |
Positivo/Dim |
HLA-DR |
Receptor de IL-2, células T y B activadas, células peludas |
Negativo |
Interpretación: Aspirado de médula ósea con perfil inmunofenotipo consistente con Leucemia Promielocítica Aguda (LPA). PML/RARA positivo por FISH. |
||
Fuente: Citometría de flujo realizada sobre aspirado medular. Datos utilizados con autorización de la paciente y familiares, conforme a principios éticos de confidencialidad.
Tabla 3. Reporte de Oncopatología Molecular. Análisis del conjunto de sondas FISH PML/RAR (Análisis morfométrico/FISH/Manual)
Cromosoma |
Ensayos con sondas |
Parámetros de detección |
Resultados |
Nomenclatura ISCN |
15q22 17q21.1 |
XL PML/RARA PLUS Translocación doble Fusión MetaSistema #D-5086-100-OG |
Translocación (15;17) |
60% / Positivo para translocación |
nuc ish (PML x 3), (RARA x 3), (PML con RARA x 2) [120/200] |
Fuente: Estudio realizado sobre aspirado de médula ósea mediante técnica FISH con sonda dual de ruptura para PML/RARa. Publicación autorizada por la paciente y familiares, conforme a principios éticos de confidencialidad y consentimiento informado.
Tabla 4. Reporte de Oncopatología Molecular. FISH para ETO/AML1 (Análisis morfométrico/FISH/Manual)
Cromosoma |
Ensayos con sondas |
Parámetros de detección |
Resultados |
Nomenclatura ISCN |
8q21.3-22.1 21q22.1 |
XL ETO/AML 1 plus Sonda de Fusión Dual para Translocación MetaSistema #D-5114-100-OG |
Translocación (8;21) |
0% / Negativo para translocación |
nuc ish (ETO x 2), (AML x 2) [200] |
Fuente: Estudio realizado sobre aspirado de médula ósea mediante técnica FISH con sonda dual de ruptura para ETO/AML1. Publicación autorizada por la paciente y familiares, conforme a principios éticos de confidencialidad y consentimiento informado.
Tras la obtención de los resultados del aspirado de médula ósea, los cuales confirmaron el diagnóstico de una neoplasia hematológica de alto riesgo, se coordinó el traslado urgente de la paciente al Instituto Nacional del Cáncer Dr. Heriberto Pieter, ubicado en Santo Domingo, a través del Centro de Regulación de Urgencias y Emergencias (CRUE). El objetivo fue garantizar el acceso a un centro especializado en hemato-oncología para la continuidad del manejo integral.
Una vez admitida, se inició tratamiento específico dirigido a la Leucemia Promielocítica Aguda (LPA), comenzando con ácido holo-transretinoico (vesanoid) a una dosis de 10 mg por vía oral, administrado en esquema fraccionado: 4 comprimidos en la mañana y 3 en la noche, por un período planificado de 90 días. Este agente promueve la diferenciación de los promielocitos malignos, siendo esencial en la fase inicial del tratamiento.
Simultáneamente, se instauró quimioterapia de inducción basada en antraciclinas, con elección entre daunorubicina (60-90 mg/m2/día) o idarrubicina (12 mg/m2/día), ambas administradas por vía intravenosa durante 3 días consecutivos, en combinación con citarabina (100- 200 mg/m2/día) por vía intravenosa por 7 días, conforme a protocolos internacionales de manejo para LPA.
Aun con la implementación temprana y protocolizada del tratamiento específico, así como del esfuerzo sostenido de un equipo médico multidisciplinario altamente comprometido, la evolución clínica de la paciente fue marcadamente desfavorable. Desde su ingreso, se estableció una vigilancia clínica estrecha, con monitoreo continuo de signos vitales, parámetros hematológicos y metabólicos, instaurándose medidas de soporte integral, incluyendo hidratación intensiva, profilaxis para complicaciones infecciosas y manejo transfusional individualizado, a fin de estabilizar su condición general antes y durante el inicio del tratamiento.
A pesar de la respuesta rápida del personal médico, del cumplimiento riguroso de las guías clínicas internacionales y de la implementación oportuna de medidas terapéuticas y de soporte, la paciente desarrollo complicaciones graves inherentes a la biología agresiva de la enfermedad, incluyendo probable síndrome de diferenciación y falla multiorgánica progresiva, que lamentablemente no respondieron a las intervenciones instauradas.
El deceso de la paciente se produjo en un entorno hospitalario controlado, luego de haberse agotado todos los recursos disponibles con el objetivo de preservar la vida, en el marco de una atención medica técnica, ética y humana de la más alta calidad.
Discusión
El presente caso representa el primer diagnóstico documentado de Leucemia Promielocítica Aguda (LPA) con reordenamicneto PML/RARA y compromiso del Sistema Nervioso Central del Hospital Regional Universitario San Vicente de Paúl, desde la instauración del programa de residencia en Medicina Interna. Adquiriendo relevancia clínica no solo por su singularidad institucional, sino también por la rareza que representa la afectación extramedular al momento del diagnóstico en esta entidad oncohematológica.
La LPA constituye una urgencia hematológica caracterizada por la translocación recíproca t(15;17) (q24;q21), la cual genera el gen de fusión PML-RARA, interfiriendo con la maduración normal de los promielocitos. Es una forma de Leucemia Mieloide Aguda con una evolución frecuentemente fulminante si no se instaura tratamiento de forma inmediata. Su presentación clínica suele acompañarse de coagulopatía severa y alto riesgo de hemorragias fatales, lo que justifica la necesidad de diagnósticos y tratamiento emergente12.
Si bien la infiltración extramedular puede observarse en fases de recaída, su aparición en el momento del diagnóstico es un hallazgo raro, y se asocia con un pronóstico más sombrío. Entre los sitios más frecuentemente afectados por esta diseminación atípica se encuentra la piel y el sistema nervioso central, siendo este último especialmente desafiante por las implicaciones clínicas, el riesgo neurológico asociado y la complejidad del manejo terapéutico12.
En este caso, la sospecha de compromiso neurológico fue clínicamente fundada por la aparición de un cuadro de déficit motor progresivo en miembros inferiores, confirmado mediante estudios tomográficos y respaldado finalmente por los hallazgos citomorfológicos y moleculares del aspirado de médula ósea, incluyendo inmunofenotipificación compatible con LPA y detección positiva del gen de fusión PML/RARA por técnicas de biología molecular (FISH).
El manejo de la infiltración al Sistema Nervioso Central en el contexto de LPA requiere un enfoque terapéutico combinado. La estrategia incluye terapia local, como la administración de quimioterapia intratecal (habitualmente con triple esquema: citarabina, metotrexato e hidrocortisona) o, en algunos casos seleccionados, irradiación craneal y terapia sistémica dirigida al control de la enfermedad de base, basada en la administración de ácido holo-transretinoico (ATRA) y agentes quimioterapéuticos como antraciclinas y citarabina, siguiendo los protocolos adaptados al riesgo12.
Este caso resalta no solo una presentación infrecuente de LPA, sino también la capacidad diagnóstica, clínica y científica desarrollada dentro de nuestra institución, gracias al fortalecimiento progresivo de los servicios de Medicina Interna, hematología y otras especialidades. Además, pone de relieve la importancia de mantener un alto índice de sospecha clínica ante manifestaciones neurológicas atípicas en paciente con síndrome hematológicos agudos, así como la necesidad de una respuesta médica precoz, coordinada y basa en la evidencia, para optimizar los resultados en condiciones de alta letalidad como la LPA.
Existen evidencias de otras manifestaciones neurológicas inusuales de esta entidad como es la presión intracraneana idiopática (PIC), la cual es infrecuente y poco descrita en la literatura. Estando relacionado su aparición con fenómenos proinflamatorios sistémicos, trombosis del sistema venos cerebral o como efecto secundario de la terapia con ácido trans-retinoico (ATRA)13. De igual manera, el sarcoma promielocitico espinal puede presentarse en fases iniciales de la LPA, asociándose a recaídas o formas agresivas de la enfermedad. En estos pacientes, el compromiso del sistema nervioso central, particularmente a nivel medular, plantea un desafío diagnóstico, dado que puede simular procesos infecciosos, inflamatorios o tumorales primarios. Estas alteraciones neurológicas inesperadas enfatizan la complejidad y agresividad potencial de la Leucemia Promielocitica Aguda, una entidad que, a pesar de su alta tasa de curación presenta manifestaciones amenazantes para la vida en donde reconocerlas no solo evita retrasos en el tratamiento, sino que también reafirma la necesidad de individualizar cada caso dentro de una enfermedad que, aunque bien caracterizada, continúa revelando nuevos desafíos clínicos14.
Conclusión
El caso descrito constituye una presentación inusualmente agresiva de Leucemia Promielocítica Aguda (LPA) con translocación PML/RARA positiva y compromiso del sistema nervioso central desde el debut clínico, un patrón poco frecuente en la literatura médica actual. Esta variante de LPA representa un reto diagnóstico y terapéutico significativo, no solo por su potencial de evolución fulminante, sino también por la complejidad que conlleva su manifestación extramedular especialmente a nivel neurológico15.
A pesar de haber sido identificada de manera oportuna, estabilizada en un entorno hospitalario con recursos diagnósticos adecuados y, posteriormente traslada para recibir tratamiento según las guías internaciones – incluyendo la administración de ácido holo-transretinoico (ATRA) y quimioterapia antileucémica –, la paciente desarrolló un curso clínico rápidamente progresivo, con aparición de severas complicaciones que no respondieron al tratamiento instaurado.
Este desenlace adverso destaca la letalidad potencial de la LPA en fases iniciales, particularmente cuando se acompaña de hiperleucocitosis y se complica con el síndrome de diferenciación inducido por ATRA, cuadro que puede evolucionar con falla multiorgánica y muerte, aun bajo manejo intensivo.
El presente caso resalta la necesidad de mantener un alto grado de sospecha clínica ante cuadros hematológicos con manifestaciones neurológicas agudas, así como la importancia de la abordaje integral, precoz y multidisciplinario, que integre no solo el tratamiento dirigido a la enfermedad de base, sino también el reconocimiento y manejo activo de las complicaciones asociadas al tratamiento, como el síndrome del ATRA.
Finalmente, esta experiencia clínica fortalece el llamado a desarrollar protocolos de acción rápida, mejorar la disponibilidad de recursos terapéuticos y consolidar la capacitación en enfermedades hematológicas de alta letalidad, con el fin de optimizar los resultados clínicos en contextos donde cada hora puede ser determinante para la supervivencia.
Recomendaciones
A partir del análisis clínico y evolutivo del presente caso, se recomienda enfáticamente que, ante la presencia de pacientes con manifestaciones hemorrágicas cutáneas inusuales como equimosis extensas o de distribución atípica, acompañadas de alteraciones hematológicas como leucocitosis, anemia, neutropenia y trombocitopenia, se considere de manera temprana dentro del diagnósticos diferencial la posibilidad de una Leucemia Promielocítica Aguda (LPA), incluso si no existen antecedentes hematológicos previos.
Dado el carácter fulminante que puede adquirir esa entidad, especialmente cuando se asocia a complicaciones como el síndrome de diferenciación inducido por el tratamiento o afectación del sistema nervioso central, se debe actuar con un alto índice de sospecha clínica y con extrema celeridad en el abordaje diagnóstico, iniciando estudios como el frotis de sangre periférica, aspirado de médula ósea y pruebas de inmonofenotipificación, sin esperar la evolución completa del cuadro.
De igual manera, se recomienda establecer canales de referencia directa y protocolos de actuación urgente con centros especializados en hematología oncológica para garantizar el inicio precoz de terapias dirigidas, como el ácido holo-transretinoico (ATRA), cuyo retraso puede impactar negativamente en la sobrevida de los pacientes.
Finalmente, es crucial fomentar la educación médica continua sobre leucemia agudas y sus presentaciones atípicas entre médicos generales, internistas, emergenciólogos y demás especialistas, con el fin de reconocer tempranamente signos de alarma en escenarios clínicos no convencionales, lo cual puede marcar la diferencia entre una evolución favorable o un desenlace fatal.
Agradecimientos
A la Dra. Leidy de Jesús, Hematóloga – Internista, por su invaluable compromiso y acompañamiento clínico en todas las fases del abordaje de esta paciente. Su participación fue fundamental tanto en el proceso diagnóstico como en la instauración del tratamiento inicial, así como en la articulación y coordinación del traslado hacia aun centro Oncológico de mayor complejidad. Asimismo, le expreso mi más profundo agradecimiento por su orientación experta y supervisión académica durante la elaboración y revisión del presente caso clínico, cuyo desarrollo no habría sido posible sin su entrega profesional y humana.
Financiamiento
Esta investigación fue desarrollada de manera independiente y no contó con el respaldo financiero de instituciones públicas, privadas ni organizaciones sin fines de lucro. Todas las actividades relacionadas con esta la investigación, análisis clínico y redacción fueron realizadas sin financiamiento externo.
Conflicto de intereses
El autor declara que no existen conflictos de intereses de carácter financiero, personal, académico ni profesional que puedan haber influido de manera inapropiada en la elaboración, análisis o publicación descrita.
Consentimiento informado
La información clínica, imágenes diagnósticas y resultados paraclínicos presentados en este caso son utilizados con fines exclusivamente académicos y científicos. La paciente y sus familiares otorgaron consentimiento informado para la recopilación y publicación de los datos, garantizando el respeto a la privacidad y confidencialidad, conforme a los principios éticos establecidos en la Declaración de Helsinki.
Referencias bibliográficas
1. Mejía Maggi Nataly Carolina, Guizado Infante Víctor Mauricio, Montenegro Corrales Carlos Arnulfo. Leucemia promielocítica aguda M3. Reporte de un caso clínico. Rev Eug Esp [Internet]. 2019 dic [citado 2024 mayo 02]; 13(2): 62-70. Disponible en: http://scielo.senescyt.gob.ec/scielo.php?script=sci_arttext&pid=S2661-67422019000200062&lng=es. https://doi.org/10.37135/ee.004.07.07
2. Mejía-Buriticá Leonardo, Torres-Hernández José Domingo, Vásquez Gonzalo de Jesús. Leucemia promielocítica aguda. Estado del arte. Iatreia [Internet]. marzo de 2021 [consultado el 2 de mayo de 2024]; 34(1): 42-53. Disponible en: http://www.scielo.org.co/scielo.php?script=sci_arttext&pid=S0121-07932021000100042&lng=en. https://doi.org/10.17533/udea.iatreia.76.
3. Cingam SR, Koshy NV. Leucemia promielocítica aguda. [Actualizado el 26 de junio de 2023]. En: StatPearls [Internet]. Isla del Tesoro (FL): StatPearls Publishing; 2024 enero-. Disponible en: https://www.ncbi.nlm.nih.gov/books/NBK459352/
4. Figueroa R, Heller V, Alonso C, Savio V, Albiero A, Yorio M, Gobbi C, Alba P. SAF y leucemia promielocítica aguda: un desafío terapéutico. Rev Fac Cien Med Univ Nac Córdoba [Internet]. 26 de octubre de 2022 [citado 3 de mayo de 2024];79(Suplemento JIC XXIII). Disponible en: https://revistas.unc.edu.ar/index.php/med/article/view/39037
5. Daniel E, Jackeline M, José H, Claudio F, Marco V, Silvia N, et al. Factores de riesgo asociados a la mortalidad muy temprana en leucemia promielocítica aguda en el Instituto Nacional de Enfermedades Neoplásicas [Internet]. Com.mx. [citado el 3 de mayo de 2024]. Disponible en: https://www.imbiomed.com.mx/articulo.php?id=113161
6. Yilmaz, M., Kantarjian, H. y Ravandi, F. Algoritmos actuales de tratamiento de la leucemia promielocítica aguda. Blood Cancer J. 11, 123 (2021). https://doi.org/10.1038/s41408-021-00514-3
7. Zheng PZ, Wang KK, Zhang QY, Huang QH, Du YZ, Zhang QH, et al. Análisis de sistemas del transcriptoma y el proteoma en la diferenciación celular/apoptosis inducida por ácido retinoico/trióxido de arsénico en la leucemia promielocítica. Proc Natl Acad Sci USA. 2005;102:7653–8.
8. Estey E, Garcia-Manero G, Ferrajoli A, Faderl S, Verstovsek S, Jones D, et al. Uso de ácido transretinoico más trióxido de arsénico como alternativa a la quimioterapia en la leucemia promielocítica aguda no tratada. Blood. 2006;107:3469–73.
9. Wang G, Li W, Cui J, Gao S, Yao C, Jiang Z, et al. Un enfoque terapéutico eficaz para pacientes con leucemia promielocítica aguda mediante una combinación de trióxido de arsénico con ácido retinoico all-trans en dosis bajas. Hematol Oncol. 2004;22:63–71.
10. Wang H.-Y., Gong S., Li G.-H., Yao Y.-Z., Zheng Y.-S., Lu X.-H., Wei S.-H., Qin W.-W., Liu H.-B., Wang M.-C., et al. Una estrategia eficaz y sin quimioterapia con ácido transretinoico y trióxido de arsénico para la leucemia promielocítica aguda en todos los grupos de riesgo (ensayo APL15). Blood Cancer J. 2022;12:158. doi: 10.1038/s41408-022-00753-y.
11. Ghiaur A, Doran C, Gaman MA, Ionescu B, Tatic A, Cirstea M, Stancioaica MC, Hirjan R, Coriu D. Acute Promyelocytic Leukemia: Review of Complications Related to All-Trans Retinoic Acid and Arsenic Trioxide Therapy. Cancers (Basel). 2024 Mar 15;16(6):1160. doi: 10.3390/cancers16061160. PMID: 38539495; PMCID: PMC10969096.
12. González Hobecker MF, Asson CS, Stemberg E, Bernard H, Fernández C. Leucemia promielocítica aguda con compromiso de sistema nervioso central: a propósito de un caso. RH [Internet]. 31 de agosto de 2020 [citado 8 de mayo de 2024];24(2):69-72. Disponible en: https://mail.revistahematologia.com.ar/index.php/Revista/article/view/268
13. Mirhosseini A, Shariat M, Ebrahimi M, Saeedy N, Nouri B. Beyond the Surface: A Unique Presentation of Acute Promyelocytic Leukemia as Idiopathic Intracranial Hypertension: A Case Report. Cancer Rep (Hoboken). 2024 Nov;7(11):e70064. doi: 10.1002/cnr2.70064. PMID: 39589180; PMCID: PMC11590332.
14. Villaseñor-Ledezma J, Amosa-Delgado M, Ruíz-Ginés JA, Álvarez-Salgado JA. Spinal promyelocytic sarcoma as the initial manifestation of acute promyelocytic leukaemia. Neurologia (Engl Ed). 2018 Oct;33(8):558-560. English, Spanish. doi: 10.1016/j.nrl.2017.02.007. Epub 2017 Apr 18. PMID: 28431836
15. Tiago VP, Cunali VCA. Síndrome de diferenciación en leucemia promielocítica aguda: relato de un caso pediátrico en el HC-UFTM. Pediatra residente. 2015;5(3):135-138
_______________________________
1 Residente de la especialidad en Medicina Interna. Hospital Regional Universitario San Vicente de Paúl, San Francisco de Macorís, República Dominicana. ORCID: https://orcid.org/0009-0009-2837-0058, email: leoneljcc5@gmail.com