Ciencia y Salud, Vol. 8, No. 1, enero-marzo, 2024 • ISSN (impreso): 0378-7680 • ISSN (en línea): 2613-8751 • Sitio web: https://revistas.intec.edu.do/
PERFIL CLÍNICO Y ELECTROMIOGRÁFICO DE LAS DISTROFIAS MUSCULARES EN REPÚBLICA DOMINCANA
Clinical and electromyographic profile of muscular dystrophies in the Dominican Republic
DOI: https://doi.org/10.22206/cysa.2024.v8i1.3057
Freddy De León Roa1, Yaneris Cesarina Polanco Melo2, Demian Herrera3, Andreina Moreno Reyes4
1 Departamento de Neurología, Hospital Infantil Dr Robert Reid Cabral, República Dominicana. Email: freddydlr0881@gmail.com
2 Departamento de Neurología, Hospital Infantil Dr Robert Reid Cabral, República Dominicana. Email: ycpm.05@gmail.com
3 Centro de Investigación Dr. Hugo Mendoza, República Dominicana. Email: herreramorbanmd@gmail.com
4 Centro de Investigación Dr. Hugo Mendoza, Hospital Pediátrico Dr. Hugo Mendoza, República Dominicana. Email: andreinamorenor3@gmail.com
Recibido: 1 de marzo, 2023 • Aprobado: 7 de febrero, 2024

Cómo citar: León Roa F. D., Polanco Melo Y. D., Herrera D., & Moreno Reyes A. (2024). Perfil clínico y electromiográfico de las distrofias musculares en República Domincana. Ciencia y Salud, 8(1), 49–56. https://doi.org/10.22206/cysa.2024.v8i1.3057
Resumen
Introducción: Las distrofias musculares son trastornos miogénicos hereditarios caracterizados por una atrofia muscular progresiva y una debilidad de distribución y gravedad variable. La población de Republica Dominicana es fruto de una mezcla de etnias, haciéndola portadora de una herencia cromosómica y ADN diverso, siendo susceptibles a poder presentar cualquier desorden de carácter hereditario.
Material y métodos: Con una muestra de 17 pacientes obtenidos entre septiembre 2019- marzo 2020, se realizó un estudio retrospectivo, descriptivo y transversal, en el cual se hizo una revisión de los expedientes de la clínica de miopatías en la consulta de neurología pediátrica del Hospital Infantil Doctor Robert Reid Cabral, para describir el perfil clínico de los pacientes con distrofia muscular y los hallazgos de electromiografía en los casos que la misma.
Resultados: se encontró que la distribución de la edad correspondió a 5-9 años en un 53%, siendo el sexo masculino, el más frecuente. En el 70.59% presentaron antecedentes familiares de distrofia muscular. Los principales motivos de consulta fueron cansancio y caídas frecuentes.
Conclusión: En los hallazgos de electromiografía, el porcentaje de pacientes que presentó esta prueba con alteraciones fue de 88.24% y sin alteraciones el 11.76%. Esto nos demuestra, la gran utilidad de dicho estudio en el diagnóstico de las distrofias musculares en países donde no se cuenta con estudio molecular, siendo una de las pruebas esenciales en el abordaje diagnóstico de los pacientes con sospecha clínica de dichas patologías.
Palabras clave: Distrofia muscular, electromiografía, prednisona.
Abstract
Introduction: Muscular dystrophies are hereditary myogenic disorders characterized by progressive muscular atrophy and weakness of variable distribution and severity. The population of the Dominican Republic is the result of a mixture of ethnic groups, making it the bearer of a diverse chromosomal inheritance and DNA, being susceptible to presenting any hereditary disorder.
Methods: With a sample of 17 patients obtained between September 2019-March 2020, a retrospective, descriptive and cross-sectional study, in which a review of the files of the myopathies clinic was made in the pediatric neurology consultation of the Children's Hospital Doctor Robert Reid Cabral, to describe the clinical profile of patients with muscular dystrophy and the electromyography findings in the cases with the same.
Results: The age distribution corresponded to 5-9 years; 53%, being the masculines, the most frequent sex. In 70.59%, there was a family history of muscular dystrophy. The main reasons for consultation were fatigue and frequent falls.
Conclusion: In the electromyography findings, the percentage of patients who presented this test with alterations was 88.24% and 11.76% without alterations. This result shows us the great utility of said study in the workup of muscular dystrophies in countries with no availabilities for molecular studies, being one of the essential tests in the diagnostic approach of patients with clinical suspicion of said pathologies.
Keywords: Muscular dystrophy, electromyography, prednisone.
Introducción
Las distrofias son un grupo de trastornos hereditarios que provocan debilidad muscular y pérdida del tejido muscular, las cuales empeoran con el tiempo, las misma están contenidas dentro de un grupo mayor de patologías conocidas como miopatías ya que comprometen el musculo estriado en forma primaria, excluyendo cualquier otra enfermedad que se origine en la unidad motora (neurona motora, axón o unión neuromuscular)1, 2.
Las distrofias musculares (DM) son un grupo de afecciones hereditarias (enfermedades genéticas), lo cual significa que se transmiten de padres a hijos. Son un grupo de más de 30 que causan debilidad y degeneración progresivas de los músculos esqueléticos usados durante el movimiento voluntario. Estos trastornos varían en la edad de inicio, gravedad, y patrón de músculos afectados. Todas las formas de distrofia muscular empeoran a medida que los músculos degeneran y se debilitan progresivamente. La mayoría de los pacientes finalmente pierde la capacidad de caminar3.
Son más o menos raras dependiendo del tipo específico. La distrofia muscular de Duchenne (DMD) es la distrofia muscular más frecuente (1 para cada 3,500 varones que nacen), la segunda es la distrofia de Becker (DMB) (1 para cada 30,000 varones que nacen). Los otros tipos son más raros (por ejemplo, la distrofia de cinturas ocurre en solo 1.3% de los pacientes con distrofia muscular). El tipo oculofaringeo es más común en Francia y Canadá y el tipo de distrofia muscular distal es más común en Suecia4.
No se sabe con exactitud cuántas personas de todas las edades tienen distrofia muscular de Duchenne o Becker en los Estados Unidos, pero se estima que sean de 1 de cada 5600 a 7700 hombres de 5 a 24 años. Esta cifra es aproximadamente igual a la prevalencia de 1.3 a 1.8 por cada 10 000 hombres de 5 a 24 años5, 6.
La población de Republica Dominicana es fruto de una mezcla de etnias, haciéndola portadora de una herencia cromosómica y ADN diverso, siendo susceptibles a poder presentar cualquier desorden de carácter hereditario.
Material y métodos
Se realizó un estudio retrospectivo, descriptivo y transversal. En el cual se hizo una revisión de los expedientes archivados y localizados de la clínica de miopatías en la consulta de neurología pediátrica para describir el perfil clínico de los pacientes con distrofia muscular y con hallazgos positivos de electromiografía. La muestra estuvo conformada por 17 pacientes atendidos en el departamento de Neurología del Hospital Infantil Dr. Robert Reid Cabral con diagnóstico o sospecha de distrofia muscular. Se excluyó a los pacientes con electromiografía no realizada. Los resultados fueron tabulados con programas informáticos del paquete Microsoft Office tales como: Excel, Word y analizados en tablas de frecuencia simple. El proyecto fue sometido y aprobado por el comité de investigación del Hospital Infantil Robert Reid Cabral.
Resultados
En nuestro estudio podemos observar que el rango de edad más frecuente en el cual los pacientes eran diagnosticados o acudían a la consulta de neurología correspondía a 5-9 años para el 53% y 10-14 años para el 35%. El sexo más afectado fue el masculino con un 76.47%. El 23.53% restante fueron mujeres (ver cuadro 1).
Cuadro 1. Diagnostico no molecular en pacientes con distrofia muscular, perfil clínico y de electromiografía, consulta neurología hospital infantil Dr. Robert Reid Cabral, septiembre 2019- marzo 2020. n=17
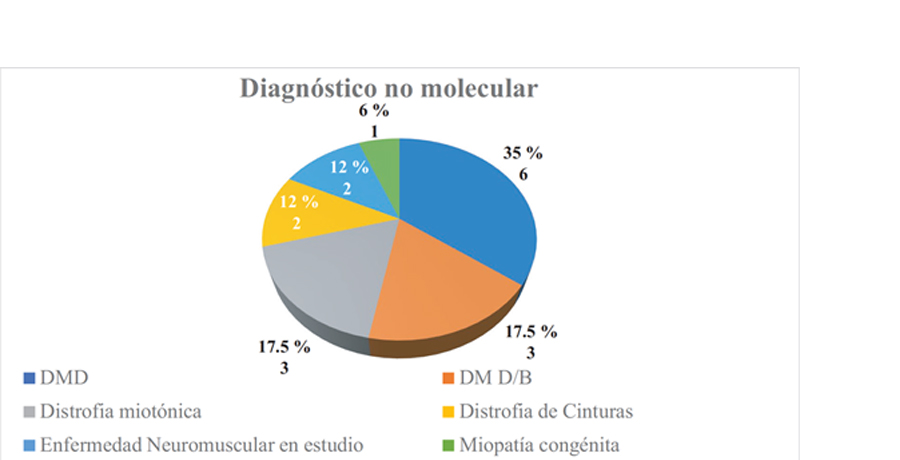
Los pacientes con antecedentes familiares representaron el 29.41% y sin antecedentes familiares el 70.59%, al considerarse una enfermedad letal recesiva ligada al cromosoma X (ver cuadro 1).
Dentro de los motivos de consulta las caídas frecuentes y el cansancio representaron el 88% y el dolor muscular se cuantificó en un 24%, estos datos se corresponden con las estadísticas de las distrofias en sentido general en comparación con estudios internacionales (ver cuadro 1).
Dentro de las manifestaciones clínicas la valoración de la fuerza muscular evidencia una disminución de 3/5 en el 64.71%, 2/5 en el 23.53% y 4-/5 en el 11.76%, un hallazgo frecuente en grados variables, pero siempre presente. Los REM se encontraban disminuidos en el 71% y ausentes en el 29% de los pacientes. La alteración de la marcha presente en el 100% de los pacientes. El tono muscular, hipotónico en el 47% de los pacientes, retraso motor en el 53% y disfagia en el 6%.
Dentro de nuestro estudio la CPK fue dividida en: rango de distrofia, no distrofia y no realizada, partiendo como rango de distrofia, valores por encima de 4 veces el valor normal como se clasifica para DMD, donde el 52.94% de los pacientes se encontraba en este renglón, el 29.41% no distrofia y el 17.65% no realizó la prueba. La LDH fue encontrada en el 23.53% a resultados alterados; el 11.76% normales; el 64.71% no realizó dicha prueba. La aldolasa se toma como referencia un rango > 8 como alterada, < 8 normal y no realizada. Para rango > 8 tenemos un 47%; no realizada tenemos 53%, dicho resultado nos arroja que todos los pacientes que se realizaron la prueba tuvieron alteraciones. En los hallazgos de electromiografía, el porcentaje de pacientes que presentó esta prueba con alteraciones fue de 88.24% y sin alteraciones el 11.76% (ver tabla 1).
Tabla 1. Perfil demográfico y clínico en pacientes con distrofia muscular, perfil clínico y de electromiografía, consulta neurología Hospital Infantil Dr. Robert Reid Cabral, septiembre 2019-marzo 2020
|
Frecuencia |
Porcentaje |
Edades |
|
|
1-4 años |
2 |
12 |
5-9 años |
9 |
53 |
10-14 años |
6 |
35 |
Sexo |
|
|
Masculino |
13 |
76.47% |
Femenino |
4 |
23.53% |
Antecedentes Familiares |
|
|
Con |
5 |
29.41% |
Sin |
12 |
70.59% |
Manifestaciones clínicas |
|
|
Motivo de Consulta |
|
|
Cansancio |
15 |
88% |
Dolor Muscular |
4 |
24% |
Caídas frecuentes |
15 |
88% |
Manifestaciones clínicas |
|
|
|
Frecuencia |
Porcentaje |
Fuerza Muscular (FM) Escala de Daniels |
|
|
4/5 FM |
2 |
12% |
3/5 FM |
11 |
65% |
2/5 FM |
4 |
24% |
Reflejos de Estiramiento Muscular (REM) |
||
Hiporreflexia |
12 |
71% |
Arreflexia |
5 |
29% |
Examen físico |
|
|
Disfagia |
1 |
6% |
Hipotonía |
8 |
47% |
Retraso Motor |
9 |
53% |
Marcha Alterada |
17 |
100% |
Electromiografía |
|
|
Con alteraciones |
15 |
0.88 |
Sin alteraciones |
2 |
0.12 |
Tratamiento |
|
|
Prednisona |
8 |
47% |
Fisioterapia/Rehabilitación |
2 |
12% |
Baclofeno |
1 |
6% |
Núcleo CMP |
1 |
6% |
Sin tratamiento |
5 |
29% |
Para el abordaje terapéutico de los pacientes con estas patologías nos enfocamos en los parámetros de manejo establecidos en las guías de manejo estandarizadas de aceptación internacional, los pacientes en el 47% de los casos fueron manejados con esteroides siendo la prednisona el utilizado, un 12% recibieron fisioterapia. Cabe mencionar que, debido al carácter retrospectivo, no se incluyó algunas variables de interés, como si el paciente caminaba al momento del inicio del esteroide, dato a tomar en cuenta en futuras investigaciones.
Discusión
En nuestro estudio podemos observar que el rango de edad más frecuente en el cual los pacientes eran diagnosticados o acudían a la consulta de neurología correspondía a 5-9 años para el 53% a diferencia de la media en todos los casos de distrofias musculares predominantes en niños donde se hacía el diagnóstico antes de los 5 años7 de edad como es el caso de la DMD o DMB8, coincidiendo con nuestro estudio con el estudio de casos de Guapi Nauñay4 en el cual la edad de los pacientes fue de 7 y 9 años. Es necesario contar con más neurólogos expertos en enfermedades neuromusculares en nuestro país, para que diagnostique e identifique más temprano los datos clínicos de distrofia muscular.
El sexo más afectado fue el masculino con un 76.47%, guardando relación con el comportamiento de las distrofias más frecuentes las cuales son ligadas al cromosoma X8, 9. El 23.53% restante fueron mujeres.
Los pacientes con antecedentes familiares representaron el 29.41% y sin antecedentes familiares el 70.59%. Al considerarse una enfermedad letal recesiva ligada al cromosoma X; por ende, el riesgo de recurrencia en una mujer portadora de la enfermedad en cada gestación es en el 50 % de hijos enfermos; en el 50 % de hijos sanos; en el 50 % de hijas portadoras y en el 50 % de hijas no portadoras. El llegar a un diagnóstico en determinada familia crea la necesidad de identificar a mujeres portadoras con la finalidad de establecer el adecuado asesoramiento genético y más ahora en donde los estudios moleculares son la elección diagnostica.
Dentro de los motivos de consulta las caídas frecuentes y el cansancio representaron el 88% y el dolor muscular se cuantificó en un 24%, estos datos se corresponden con las estadísticas de las distrofias en sentido general en comparación con estudios internacionales8-14. Lo cual coincide con el estudio de Esquivel5 en el cual el motivo de consulta más frecuente fue la dificultad para subir gradas.
Dentro de las manifestaciones clínicas la valoración de la fuerza muscular evidencia una disminución de 3/5 en el 64.71%, 2/5 en el 23.53% y 4-/5 en el 11.76%, un hallazgo frecuente en grados variables, pero siempre presente. Los REM se encontraban disminuidos en el 71% y ausentes en el 29% de los pacientes. La alteración de la marcha presente en el 100% de los pacientes. El tono muscular, hipotónico en el 47% de los pacientes, retraso motor en el 53% y disfagia en el 6%. Esto coincide con el estudio de Esquievel5 en el cual el hallazgo al examen físico más frecuente fue la pseudohipertrofia de gastrocnemios seguido de la debilidad muscular en miembros inferiores. En esta investigación no mencionamos la afección cardiovascular, por no ser un objetivo a estudiar. No se hace referencia a las alteraciones cognoscitivas ya que la mayoría de los pacientes no contaban con análisis específicos de coeficiente intelectual o no asisten a escuelas.
Dentro de nuestro estudio la CPK fue dividida en: rango de distrofia, no distrofia y no realizada, partiendo como rango de distrofia, valores por encima de 4 veces el valor normal como se clasifica para DMD, donde el 52.94% de los pacientes se encontraba en este renglón, el 29.41% no distrofia y el 17.65% no realizó la prueba. La LDH fue encontrada en el 23.53% a resultados alterados; el 11.76% normales; el 64.71% no realizó dicha prueba. La aldolasa se toma como referencia un rango > 8 como alterada, < 8 normal y no realizada. Para rango > 8 tenemos un 47%; no realizada tenemos 53%, dicho resultado nos arroja que todos los pacientes que se realizaron la prueba tuvieron alteraciones, el resto de pacientes no realizó la misma y por eso, no tenemos ningún resultado normal, aunque podemos asumir por la tendencia de resultados que de haber realizado dicha prueba todos los pacientes habrían tenido resultados alterados.
En los hallazgos de electromiografía, el porcentaje de pacientes que presentó esta prueba con alteraciones fue de 88.24% y sin alteraciones el 11.76%. Esto nos demuestra, la gran utilidad de dicho estudio en el diagnóstico de las distrofias musculares en países donde no se cuenta con estudio molecular, siendo una de las pruebas esenciales en el abordaje diagnóstico de los pacientes con sospecha clínica de dichas patologías14, 16-20. Coincidiendo con el estudio de Guapi Nauñay4. Sin embargo, de este aspecto se proponen estudios prospectivos posteriores cuando contemos en la institución con estudio de EMG.
Dentro de los diagnósticos de distrofia muscular que encontramos en nuestro estudio, el diagnóstico más frecuente fue de distrofinopatías donde la distrofia muscular de Duchenne obtuvo un 35.29%, seguido de la DM D/B para un 17.65% y la Distrofia miotónica para un 17.65%.
Para el abordaje terapéutico de los pacientes con estas patologías nos enfocamos en los parámetros de manejo establecidos en las guías de manejo estandarizadas de aceptación internacional, los pacientes en el 47% de los casos fueron manejados con esteroides siendo la prednisona el utilizado, un 12% recibieron fisioterapia. Cabe mencionar que, debido al carácter retrospectivo, no se incluyó algunas variables de interés, como si el paciente caminaba al momento del inicio del esteroide, dato a tomar en cuente en futuras investigaciones. Esto coincide con el estudio de Guapi Nauñay4 en el cual los pacientes fueron tratados con esteroides, pero no descartamos que pronto podamos tener nuevos protocolos donde reclutemos a pacientes con sospecha de la enfermedad que puedan caminar y que sean fuertes candidatos para la terapia génica, desde luego con confirmación por el estudio genético.
Conclusiones
A partir de los resultados llegamos a las siguientes conclusiones:
- La moda con respecto al rango etario fue de 5-9 años en los pacientes, en donde más de la media era de sexo masculino y en quienes se encontraron antecedentes familiares de distrofia muscular.
- Los principales motivos de consulta fueron cansancio y caídas frecuentes, quienes presentaron alteración de la marcha, al momento de la evaluación. Los mismos eran diagnosticados e identificados de manera tardía, considerando el factor tiempo como una variable dependiente de la gravedad de los síntomas de la enfermedad.
- En los hallazgos de laboratorio, los niveles de CPK presentaron rango de distrofia, los niveles de LDH y aldolasa se encontraban por encima de sus rangos de referencia. Por otra parte, la electromiografía presentaba un patrón miopático. Esto corrobora nuestra hipótesis de que el perfil clínico de los pacientes diagnosticados con algún tipo de distrofia se corresponde con los hallazgos de la electromiografía.
- Dentro de los diagnósticos de distrofia muscular que encontramos en nuestro estudio, el diagnóstico más frecuente fue de distrofinopatías con una mayor incidencia en la distrofia muscular de Duchenne, en quienes fue usado una medicación con prednisona.
Recomendamos al Ministerio de Salud Pública brindar el soporte económico necesario a estas familias para la realización de pruebas genéticas y demás pruebas de apoyo diagnósticos. A la Sociedad Dominicana de Pediatría dar a conocer y entender los aspectos relevantes de esta enfermedad, a fin de que sea diagnosticada precozmente; a su vez, se debe tener en consideración que existen otros tipos de distrofias musculares cuya información es necesaria para establecer el diagnóstico diferencial. Al Hospital Infantil Dr. Robert Reid Cabral, recomendamosInstruir a los residentes de pediatría y neurología pediátrica en un adecuado y detallado examen clínico que busque los signos y los síntomas propios de la enfermedad; incluir las pruebas complementarias pertinentes mencionadas en esta investigación; así como el estudio molecular de la alteración génica.
Bibliografia
1. Selcen D. Muscle diseases. In: Goldman L, Schafer AI, eds. Goldman-Cecil Medicine. 25th ed. Philadelphia, PA: Elsevier Saunders. 2016:chap 421.
2. Distrofia Muscular, National Institute of Neurological Disorders and Stroke (NINDS), última revisión 30/12/2016. Diponible en: https://espanol.ninds.nih.gov/trastornos/distrofiamuscular.htm
3. Distrofia Muscular. Datos y estadísticas. Centros para el Control y la Prevención de las Enfermedades. https://www.cdc.gov/ncbddd/spanish/musculardystrophy/data.html. Accessed 5/27/2015.
4. Guapi Nauñay VH, García Orbe JR. Distrofia muscular de Duchenne: reportes de caso. Univ Med. 2017; 58(4): 1-6. https://doi.org/10.11144/Javeriana.umed58-4.duch
5. Esquivel Zúñiga MR. Caracterización fenotípica - genotípica de los pacientes con análisis molecular por Distrofia Muscular de Duchenne y Becker en el Hospital Nacional de Niños “Dr. Carlos Sáenz Herrera” del 1 de enero del 2001 al 31de diciembre del 2017. Universidad de Costa Rica. Sistema de estudios de posgrado. 2017.
6. Figueiredo Pedrosa, F. Los aspectos cognitivos de la distrofia muscular de Duchenne: una revisión integradora. 2019. 63f. Trabajo de Conclusión del Curso (Licenciatura en Medicina) - Centro de Formación Docente, Universidad Federal de Campina Grande, Cajazeiras, Paraíba, Brasil. 2019.
7. Udd B, Krahe R, Wallgren-Pettersson C. Proximal myotonic dystrophy: a family with autosomal dominant muscular dystrophy, cataracts, hearing loss and hypogonadism: heterogeneity of proximal myotonic syndromes? Neuromuscular Disorders. 1997; 7: 217-228.
8. Johnson NE. Myotonic Muscular Dystrophies. Continuum (Minneap Minn). 2019; 25(6): 1682-1695. https://doi.org/10.1212/CON.0000000000000793 PMID: 31794466.
9. Dubowitz V. La detección de la distrofia muscular de Duchenne. Arco Dis Child. 1976; 51: 249-251.
10. Harper PS. Myotonic dystrophy, 3rd edn. London: Saunders. 2001.
11. Matthew Pitt. Neurophysiological strategies for the diagnosis of disorders of the neuromuscular junction in children. Developmental Medicine & Child Neurology. 2008; 50: 328 333. https://doi.org/10.1111/j.1469-8749.2008.02038.x
12. Madera MF, Hughes SC, Hache LP. Actitudes de los padres hacia cribado neonatal para la distrofia muscular de Duchenne / BeCPKer y atrofia muscular espinal. Muscle Nerve. 2014; 49: 822-828.
13. Hechos sobre las distrofias musculares del anillo óseo, en: https://www.mda.org/sites/default/files/publications/FactsLGMDSpanish.pdf
14. Hauser, MA, Conde, CB, Kowaljow, V. Myotilin Mutation Found in Second Pedigree with LGMD1A. The American Journal of Human Genetics. 2002; 71(6): 1428-1432. https://doi.org/10.1086/344532
15. Ashizawa T. Myotonic dystrophy as a brain disorder. Arch Neurol. 1998; 55: 291-293.
16. Darras BT, Urion DK, Ghosh PS. Dystrophinopathies. In: Adam MP, Ardinger HH, Pagon RA, et al. GeneReviews.Seattle (WA): University of Washington. 1993 2019. Available at https://www.ncbi.nlm.nih.gov/books/NBK1119.
17. Birnkrant DJ, Bushby K, Bann CM. Diagnóstico y manejo de la distrofia muscular de Duchenne, parte 1: diagnóstico y manejo neuromuscular, rehabilitación, endocrino, gastrointestinal y nutricional. Lancet Neurol. 2018; 17(3): 251 267.
18. Chung J, Smith AL, Hughes SC. Twenty-year follow-up of newborn screening for patients with muscular dystrophy. Muscle Nerve. 2016; 53: 570 578.
19. Emery A, Dreifuss, F. E. Unusual type of benign x-linked muscular dystrophy. Journal of Neurology, Neurosurgery & Psychiatry. 1966; 29(4), 338-342.
20. Pegoraro E, Hoffman E P. Limb-Girdle Muscular Dystrophy Overview. En: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews. Seattle (WA): University of Washington, Seattle. 1993-2020. August 30, 2012. https://www.ncbi.nlm.nih.gov/books/NBK1408/.