Ciencia y Salud, Vol. VII, No. 1, enero-marzo, 2023 • ISSN (impreso): 2613-8816 • ISSN (en línea): 2613-8824 • Sitio web: https://revistas.intec.edu.do/
ANEMIA APLÁSICA: UN NUEVO RETO FARMACOLÓGICO EN LA PRÁCTICA CLÍNICA
Aplastic anemia: a new pharmacological challenge in clinical practice
Escuela de Ciencias de la Salud, Facultad de Medicina, Universidad Pontificia Bolivariana, Medellín, Colombia. ORCID: 0000-0003-4044-2463, Correo-e: isabela.robledo@upb.edu.co

Cómo citar: Robledo Barrios IM. Anemia aplásica: un nuevo reto farmacológico en la práctica clínica. cysa [Internet]. 9 de marzo de 2023 [citado 9 de marzo de 2023];7(1):37-46. Disponible en: https://revistas.intec.edu.do/index.php/cisa/article/view/2771
Introducción
La anemia aplásica (AA) es una enfermedad poco frecuente caracterizada por presentar una insuficiencia en la médula ósea y una pancitopenia, sin rastro de procesos mieloproliferativos o fibróticos.1 La patogénesis resulta en la destrucción de las células hematopoyéticas, principalmente, debido a tres mecanismos: una lesión directa que afecta el microambiente estromal de la médula ósea, una producción y liberación alterada de los factores hematopoyéticos de crecimiento (inmunosupresión humoral) y un síndrome congénito o adquirido que afecta la médula ósea.2
Paul Ehrlich fue el primero en describir la enfermedad en 1888, mientras inspeccionaba a una mujer embarazada.3 Sin embargo, el término “anemia aplásica” fue acuñado por Louis Henri Vaquez, en 1904, y las manifestaciones clínicas fueron descritas por Richard C. Cabot y otros patólogos, a inicios del siglo xx.2
Lo que hace unos años era considerada una enfermedad devastadora, en la actualidad puede llegar a ser tratada, en primera medida, con un trasplante alogénico de medula ósea y, en pacientes no aptos al trasplante, se suele implementar la terapia inmunosupresora.4 Se debe tener en cuenta que el manejo terapéutico es personalizado para cada paciente, ya que depende de la etiología que presente cada uno.3
De acuerdo con los estudios epidemiológicos recientes, la estimación anual de incidencia de AA es de 2 por millón de habitantes en los países occidentales; por otro lado, en la población asiática la incidencia es alrededor de tres veces mayor.5
Tanzania es el país asiático con la mayor incidencia de casos de AA, llegando a reportar 6 casos por millón de habitantes cada año.6 En Pakistán se describe como la segunda enfermedad hematológica no maligna más común, por debajo de la Talasemia.5
La incidencia de la anemia aplásica se reporta como bifásica, debido a que muestra dos picos en los cuales se suele presentar la enfermedad; el primero se presenta entre los 15-25 años, y se ha descrito un grupo reducido después de los 60 años de edad.7 Además, la prevalencia de la AA es más alta en Asia, en donde se realizó un estudio durante 15 años, que logró asociar la presentación de la enfermedad a factores ambientales y socioeconómicos.8 Se han presentado reportes en donde se establece una predominancia de la población masculina en los casos registrados de AA.9
Un estudio sueco demostró que la mayoría de muertes en pacientes ocurren entre los dos primeros años después del diagnóstico, un 50 % es debido a infecciones graves y otro 15 % se debe a hemorragias, de ahí deriva la importancia del tratamiento temprano y la prevención de infecciones con antibióticos o terapia antimicótica.10
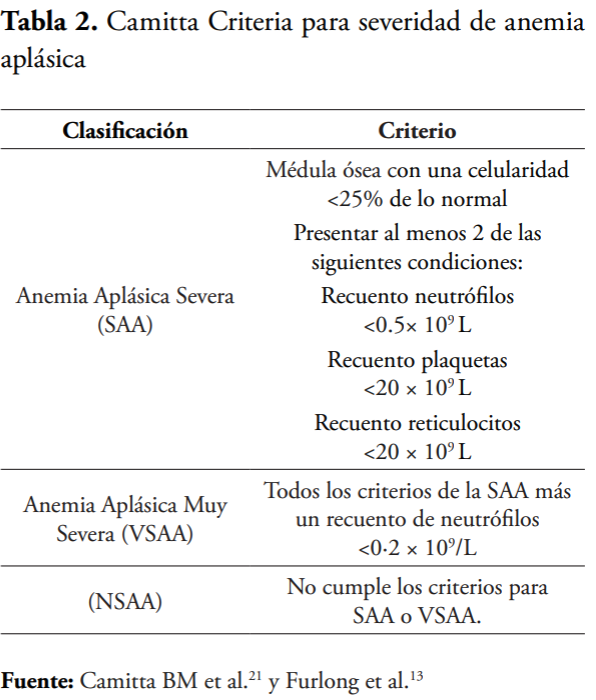
El diagnóstico de la patología se basa en realizar un aspirado o biopsia de médula ósea, a su vez se debe complementar con un recuento sanguíneo completo.11 Los pacientes diagnosticados con AA son clasificados, según su severidad, usando el criterio de Camitta.5
Aunque la anemia aplásica sea considerada un tipo de anemia no común, hoy en día continúa representando una problemática mundial, debido a que, sin tratamiento, esta enfermedad tiene una tasa de mortalidad de hasta el 70 % en los primeros dos años.11
Este artículo tiene como objetivo dar a conocer los diversos tratamientos farmacológicos usados en la terapia inmunosupresora (IST) en la AA. Adicionalmente, se profundizará en la respuesta de los pacientes frente a la IST y los mecanismos de acción de los fármacos utilizados. De igual forma, se clasifica la severidad de la AA, se mencionan algunos métodos diagnósticos y se explica la fisiopatología junto a la clínica de la enfermedad, teniendo en cuenta las diversas etiologías que se pueden presentar.
Materiales y métodos
Se realizó una búsqueda sistemática en bases de datos de literatura médica como PUBMED, British Medical Journal, New England Journal, entre otros.
Se incluyeron artículos en inglés y español; la revisión bibliografía consta de 52 referencias, desde artículos originales, artículos de revisión, reportes de caso, estudios comparativos y la guía de Camitta usada para el manejo de AA.
La estrategia de búsqueda estuvo compuesta por vocabulario controlado por el MeSH y el DeCS, se utilizó un lenguaje libre, considerando sinónimos, abreviaturas, acrónimos, variaciones ortográficas y plurales.
Clínica y fisiopatología de la enfermedad
La presentación inicial de AA se manifiesta inicialmente con fatiga, debilidad, palidez y cefalea, toda esta sintomatología es debida a la anemia.12 Posteriormente, se observa con frecuencia que los pacientes presentan petequias en piel y mucosas, además de epistaxis o gingivorragia, indicando así, una trombocitopenia.2 En un estadio más avanzado de la enfermedad, la fiebre y las infecciones suelen ser la clínica característica del paciente, debido a que ya se presenta una disminución notable de los glóbulos blancos y una neutropenia considerable.11
Se había mencionado que existían tres mecanismos para desarrollar la enfermedad:
- Daño directo: puede ser iatrogénico o dosis dependiente resultante de quimioterapia y radioterapia, también puede ser causado por medicamentos. Se manifiesta con una reducción notable en la hematopoyesis.13
- Daño inmune: numerosos estudios han mostrado que un desorden de origen inmunológico en los linfocitos T, mediado por las células madre hematopoyéticas, es el principal factor implicado en
la patogénesis de la AA.14 Adicionalmente, se ha demostrado que muchas citoquinas proinflamatorias, incluyendo el interferón gamma (IFN-γ), el factor de necrosis tumoral alfa (TNF-α), entre otras, están implicadas en el desarrollo de la enfermedad.15
- Síndromes causantes de insuficiencias en la médula ósea, ya sean congénitos o adquiridos16 (véase Tabla 1). En la mayoría, el problema es la presencia de ADN defectuoso debido a mutaciones, esto genera apoptosis y degeneración de las células madre hematopoyéticas, que, en última instancia, progresa hasta convertirse en SAA.17 Por otro lado, también se presentan enfermedades clonales, que dan lugar a patologías como la mielodisplasia, hemoglobinuria paroxística nocturna (HPN), entre otros.18
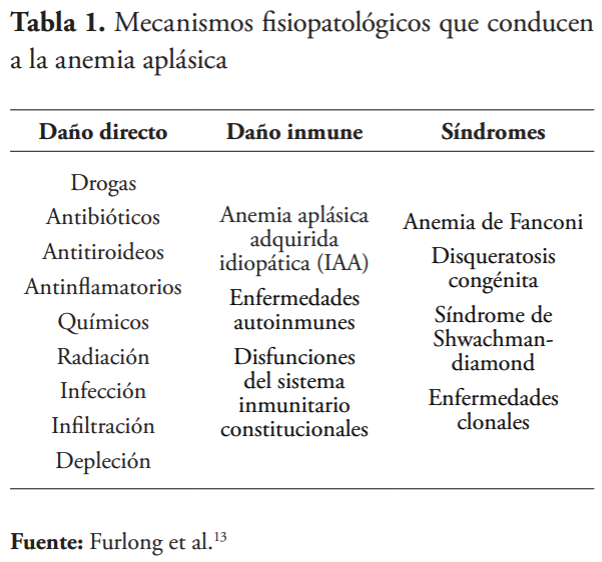
Severidad de la enfermedad
El criterio de evaluación para la severidad y el pronóstico se divide en tres:
- No severa AA (NSAA)
- Severa AA (SAA)
- Muy severa AA (VSAA)
Esta clasificación está basada según el grado de pancitopenia que se presente.19
La mayoría de las anemias aplásicas idiopáticas son consecuencia de la destrucción inmune mediada por las células hematopoyéticas, esto resulta en la presencia de diferentes grados de pancitopenia.20 Por décadas se han utilizado los criterios de Camitta (véase Tabla 2) para clasificar la gravedad de la enfermedad:
Métodos diagnósticos
El diagnóstico inicial está centrado en distinguir la AA de otras etiologías que causen pancitopenia.11
Para confirmar el diagnóstico de anemia aplásica se deben seguir estos parámetros:
- Excluir otras causas de pancitopenias o de hipocelularidad en médula ósea.
- Excluir IBMFSs (síndromes hereditarios de falla medular).
- Detectar una causa subyacente.
- Documentar posibles clones coexistentes anormales y clones de PNH (hemoglobinuria paroxística nocturna).22
El clon de PNH se presenta con frecuencia en los síndromes de falla medular adquiridos y se manifiesta en más de la mitad de los casos de AA.23
Si se confirma el diagnóstico de pancitopenia, se procede a realizar una biopsia de médula ósea, con el fin de descartar otros diagnósticos como leucemia o MDS.11 Luego, se realiza un aspirado de medula ósea, aquí se utiliza la citogenética (FISH) para descartar anormalidades cromosómicas.24 Después, se hace el recuento periférico sanguíneo para evaluar la severidad de la enfermedad13 (véase Tabla 2).
Manejo terapéutico de la anemia aplásica
Debido a la baja frecuencia de la enfermedad, las guías de tratamiento idiopático para la SAA en niños usan tanto los datos evidenciados en población adulta como pediátrica para tratar la enfermedad, llegando así a establecer el trasplante de células madres hematopoyéticas (HSCT) como tratamiento de primera línea en niños.25
Si se identifica un donante familiar compatible con HLA, el trasplante de médula debe ser la terapia de primera línea en pacientes menores de 40 años, si no se logra hallar un donante compatible, el tratamiento de elección es la terapia inmunosupresora (IST).26
Actualmente, la terapia inmunosupresora (IST) consta de globulina antitimocítica (ATG) + Ciclosporina como tratamiento estándar, debido a que en dos tercios de los pacientes tratados con estos medicamentos se ha logrado evidenciar una mejora considerable.27 Sin embargo, la eficacia de la IST está limitada por la falta de respuesta por parte de los pacientes, constantes recaídas y evolución clonal, llegando así a presentarse enfermedades como hemoglobinuria paroxística nocturna (HPN), leucemia mieloide aguda (LMA), entre otras.28
Aproximadamente el 70 % de los pacientes responden a la terapia inmunosupresora (IST) y muestran la restauración sostenida de la hematopoyesis.29 Sin embargo, entre el 5 % y el 10 % de los pacientes con AA desarrollan síndromes mielodisplásicos (MDSS) o leucemia mieloide aguda (LMA) después de un largo período de latencia, por lo que la AA se considera un estado preleucocitario similar al MDSS de menor riesgo.30
Adicionalmente, el recuperamiento tardío de las células sanguíneas periféricas expone al paciente a complicaciones tanto infecciosas como hemorrágicas, por eso es recomendada la trasfusión sanguínea y la terapia antimicótica como terapia de soporte para pacientes con SAA.31
En las últimas tres décadas, muchos estudios han demostrado el fracaso de métodos para mejorar los resultados de la IST estándar; los que fueron probados incluían la sustitución de ATG de caballo por ATG de conejo, alemtuzumab o ciclofosfamida; la adición de un tercer fármaco inmunosupresor, como el micofenolato mofetilo o el sirolimus; y la adición de factores de crecimiento hematopoyético a la terapia inmunosupresora estándar.32
En un estudio sobre la respuesta inicial frente a la globulina antitimocítica, se compararon los dos tipos de ATG presentes en el mercado: globulina antitimocítica de conejo (rATG) y de caballo (hATG), donde se reportaron respuestas inferiores de supervivencia con la rATG.33
El único país que usa la rATG como medicamento de primera línea en IST es China.34 Algunos estudios in vitro demostraron que el rATG inducía una linfopenia más grave y prolongada que el hATG, afectando, principalmente, al subconjunto de células T CD4+ que impide la recuperación hematopoyética.35
En el caso del alemtuzumab se reportó una tasa de mejora muy reducida en comparación a pacientes que usan la IST estándar;36 por otro lado, se demostró que las altas dosis de ciclofosfamida en pacientes con SAA generan, como principal complicación, una cardiotoxicidad severa.37
El micofenolato de mofetilo tiene actividad contra los linfocitos T y B autorreactivos.38 Este medicamento ha demostrado eficacia en la trombocitopenia inmune refractaria, incluso en pacientes cuya enfermedad es resistente a los glucocorticoides, lo que sugiere un mecanismo de acción complementario; pese a ello, no se reportó ningún beneficio en pacientes con AA.39
Algunos pacientes con SAA adicionaron Sirolimus a su terapia inmunosupresora. En niños se evidenció una disminución en la presentación de alteraciones clonales; no obstante, en adultos no se observó ningún beneficio.40
En algunos estudios realizados, con un número muy pequeño de casos, se evidencia una eficacia potencial en la adición de factores de crecimiento hematopoyéticos de tipo plaquetario, como lo son el Romiplostim (ROM) y eltrombopag (EPAG), en el contexto de pacientes que fueron sometidos a HSCT, el cual es el tratamiento preferencial en la AA.41
El Romiplostim es un péptido con actividad agonista de la cMPL que estimula la producción endógena de trombopoyetina, y conduce a promover la proliferación y diferenciación de los megacariocitos en la médula ósea.42
Recientemente, se ha aprobado el uso de ROM para la AA refractaria en Corea y Japón, basándose en los resultados de un ensayo internacional que demostró una respuesta positiva al uso de Romiplostim en pacientes que padecían AA refractaria, en aproximadamente un 83,9 %.43 Por el contrario, un estudio francés reveló que, aunque el tratamiento con ROM no era tóxico, en comparación con otros fármacos, muestra un beneficio muy bajo en pacientes con SAA refractaria.44
Se ha logrado evidenciar una mayor efectividad al combinar el Romiplostim con la IST estándar para obtener mejores resultados en los pacientes con SAA.45 El ROM está aprobado para el tratamiento de la púrpura trombocitopénica crónica refractaria.46
Eltrombopag es una proteína artificial que funciona como agonista del receptor de trombopoyetina (RTPO), su mecanismo de acción consiste en interactuar con el dominio transmembrana del receptor por medio de JAK-STAT, generando así una cascada de señalización.47 En estudios previos, en pacientes con anemia aplásica grave refractaria se evidencia un aumento de precursores hematopoyéticos.48
No obstante, aún no se ha estudiado en profundidad el uso de este medicamento por un período prolongado de tiempo.32 EPAG está aprobado para el tratamiento de la trombocitopenia inmunitaria refractaria y la trombocitopenia secundaria a la infección por hepatitis C.45
Se ha demostrado que la adición de eltrombopag en la terapia inmunosupresora estándar mejoró la tasa, la rapidez y la fuerza de la respuesta hematológica en los pacientes con anemia aplásica grave sin tratamiento previo, sin generar efectos tóxicos adicionales.32
En pacientes que estén próximos a someterse a un HSCT se recomienda hacer una inmunosupresión con la finalidad de evitar complicaciones, para esto se utiliza un régimen de acondicionamiento consistente en Fludarabina, dosis reducidas de Ciclofosfamida y dosis bajas de Timoglobulina.49
Otra alternativa es un trasplante de sangre de cordón umbilical, opción posible para los pacientes con anemia aplásica (AA) sin un donante emparentado o no emparentado con HLA, especialmente en pacientes donde no se evidencia mejora o respuesta frente a la terapia inmunosupresora (IST) o en pacientes donde se deba realizar el trasplante con urgencia.50
Finalmente, se considera la opción de un trasplante de donante compatible sin relación familiar; sin embargo, suele ser descartada rápidamente debido al alto riesgo de complicaciones que condicionan al paciente a una mortalidad temprana.51
Conclusiones
La anemia aplásica es una patología misteriosa, la cual representa un reto para la medicina moderna. Aunque a través del tiempo se ha logrado un gran avance en entender la fisiopatología, los métodos diagnósticos, la severidad y el manejo de la enfermedad, aún existen muchas incógnitas en cuanto al tratamiento a seguir.
En la actualidad, existen muchos fármacos, como lo son Eltrombopag y el Romiplostim, que pueden lograr convertirse en actores relevantes para un tratamiento a futuro; sin embargo, gracias a los nuevos hallazgos médicos, las alternativas terapéuticas se encuentran enfocadas cada vez más hacia la medicina personalizada, lo cual es fundamental en esta patología debido a la variedad de etiologías que presenta. Por ello, la anemia aplásica aún representa un desafío interesante para el mundo de las Ciencias de la Salud.
Referencias
- Delehaye F, Habes D, Dourthe ME, Bertrand Y, Michel G, Gaudichon J, et al. Management of childhood aplastic anemia following liver transplantation for nonviral hepatitis: A French survey. Pediatr Blood Cancer. 2020;67(4):e28177. doi: 10. 1002/pbc.28177
- Young NS. Aplastic anemia. N Engl J Med. 2018; 379:1643-56. doi: 10.1056/NEJMra1413485
- Putra A, Ngura A, Wiradewi, A. Characteristic overview of aplastic anemia at Sanglah Hospital, BaliIndonesia in 2017-2018. Intisari Sains Medis. 2019;10(3):497-500. doi: 10.15562/ism.v10i3.429
- Filippidou M, Avgerinou G, Tsipou H, Tourkantoni N, Katsibardi K, Vlachou A, et al. Longitudinal evaluation of eltrombopag in paediatric acquired severe aplastic anaemia. Br J Haematol. 2020;190(3):e157-e59. doi: 10.1111/bjh.16766
- Akram Z, Ahmed P, Kajigaya S, Satti TM, Satti HS, Chaudhary QUN, et al. Epidemiological, clinical and genetic characterization of aplastic anemia patients in Pakistan. Ann Hematol. 2019;98(2):301-12. doi: 10.1007/s00277-018-3542-z
- Ally M, Magesa P, Luzzatto L. High frequency of acquired aplastic anemia in Tanzania. Am J Hematol. 2019;94(4):E86-E88. doi: 10.1002/ajh. 25388
- John CO, Korubo K, Ogu R, Mmom CF, Mba AG, Chidiadi EA, et al. Management of aplastic anaemia in pregnancy in a resource poor centre. The Pan African Medical Journal. 2016;24:277. doi: 10.11604/pamj.2016.24.277.9880
- Ahmed P, Chaudhry QUN, Satti TM, Mahmood SK, Ghafoor T, Shahbaz N, et al. Epidemiology of aplastic anemia: a study of 1324 cases. Hematology. 2020;25(1):48-54. doi: 10.1080/ 16078454.2019.1711344
- Taj M, Shah T, Aslam SK, Zaheer S, Nawab F, Shaheen S, et al. Environmental determinants of aplastic anemia in Pakistan: a case-control study. Z Gesundh Wiss. 2016;24(5):453-60. doi: 10.1007/s10389-016-0743-6
- Vaht K, Göransson M, Carlson K, Isaksson C, Lenhoff S, Sandstedt A, et al. Incidence and outcome of acquired aplastic anemia: real-world data from patients diagnosed in Sweden from 2000-2011. Haematologica. 2017;102(10):1683-90. doi: 10.3324/haematol.2017.169862
- DeZernAE,ChurpekJE.Approachtothediagnosis of aplastic anemia. Blood Adv. 2021;5(12):2660-71. doi: 10.1182/bloodadvances.2021004345
- Dong X, Han Y, Abeysekera IR, Shao Z, Wang H. GDF11 is increased in patients with aplastic anemia. Hematology. 2019;24(1):331-6. doi: 10.10 80/16078454.2019.1574386
- Furlong E, Carter T. Aplastic anaemia: Current concepts in diagnosis and management. J Paediatr Child Health. 2020;56(7):1023-8. doi: 10.1111/jpc. 14996
- LinS,HouL,LiuS,WangJ,ChenQ,ZhangB,et al. Roles of regulatory T cells in the pathogenesis of pediatric aplastic anemia. Pediatr Hematol Oncol. 2019;36(4):198-210. doi: 10.1080/08880018.2019. 1621968
- Zhang J, Wu Q, Shi J, Ge M, Li X, Shao Y, et al. Involvement of interleukin-21 in the pathophysiology of aplastic anemia. Eur J Haematol. 2015;95(1):44-51. doi: 10.1111/ejh.12471
- WuZ,GiudiceV,ChenJ,SunW,LinZ,Keyvanfar K, et al. Interleukin-18 plays a dispensable role in murine and likely also human bone marrow failure. Exp Hematol. 2019;69:54-64.e2. doi: 10.1016/j. exphem.2018.10.003
- Ebens CL, DeFor TE, Tryon R, Wagner JE, MacMillan ML. Comparable Outcomes after HLA-Matched Sibling and Alternative Donor Hematopoietic Cell Transplantation for Children with Fanconi Anemia and Severe Aplastic Anemia. Biol Blood Marrow Transplant.2018;24(4):765-71. doi: 10.1016/j.bbmt.2017.11.031
- Cooper JP, Farah RJ, Stevenson PA, Gooley TA, Storb R, Scott BL. Hematopoietic Cell Transplantation for Paroxysmal Nocturnal Hemoglobinuria in the Age of Eculizumab. Biol Blood Marrow Transplant. 2019;25(7):1331-9. doi: 10.1016/j.bbmt. 2019.01.033
- Li Y, Ding S, Liu C, Chen T, Liu H, Li L, et al. Abnormalities of quantities and functions of CD56bright natural killer cells in non-severe aplastic Anemia. Hematology. 2019;24(1):405-12. doi: 10.1080/16078454.2019.1590963
- Giammarco S, Peffault de Latour R, Sica S, Dufour C, Socie G, Passweg J, et al. Transplant outcome for patients with acquired aplastic anemia over the age of 40: has the outcome improved. Blood. 2018;131(17):1989-92. doi: 10.1182/blood-2017-09-807859
- Camitta BM, Rappeport JM, Parkman R, Nathan DG. Selection of patients for bone marrow transplantation in severe aplastic anemia. Blood. 1975;45(3):355-63.
- Killick SB, Bown N, Cavenagh J, Dokal I, Foukaneli T, Hill A, et al. Guidelines for the diagnosis and management of adult aplastic anaemia. Br J Haematol. 2016;172(2):187-207. doi: 10.1111/bjh.13853
- Lian Y, Shi J, Nie N, Huang Z, Shao Y, Zhang J, et al. Evolution patterns of paroxysmal nocturnal hemoglobinuria clone and clinical implications in acquired bone marrow failure. Exp Hematol. 2019;77:41-50. doi: 10.1016/j.exphem.2019.08.005
- Bacigalupo A. How I treat acquired aplastic anemia. Blood. 2017 Mar 16;129(11):1428-36. doi: 10.1182/blood-2016-08-693481
- Dufour C, Veys P, Carraro E, Bhatnagar N, Pillon M, Wynn R, et al. Similar outcome of upfront-unrelated and matched sibling stem cell transplantation in idiopathic paediatric aplastic anaemia. A study on behalf of the UK Paediatric BMT Working Party, Paediatric Diseases Working Party and Severe Aplastic Anaemia Working Party of EBMT. Br J Haematol. 2015;171(4):585-94. doi: 10.1111/bjh.13614
- Rogers ZR, Nakano TA, Olson TS, Bertuch AA, Wang W, Gillio A, et al. Immunosuppressive therapy for pediatric aplastic anemia: a North American Pediatric Aplastic Anemia Consortium s10.3324/haematol.2018.206540
- Alvarado LJ, Huntsman HD, Cheng H, Townsley DM, Winkler T, Feng X, et al. Eltrombopag maintains human hematopoietic stem and progenitor cells under inflammatory conditions mediated by IFN-γ. Blood. 2019;133(19):2043-55. doi: 10.1182/ blood-2018-11-884486
- LanY,LiuF,ChangL,LiuL,ZhangY,YiM,etal. Combination of umbilical cord mesenchymal stem cells and standard immunosuppressive regimen for pediatric patients with severe aplastic anemia. BMC Pediatr. 2021;21(1):102. doi: 10.1186/s12887021-02562-x
- Winkler T, Fan X, Cooper J, Desmond R, Young DJ, Townsley DM, et al. Treatment optimization and genomic outcomes in refractory severe aplastic anemia treated with eltrombopag. Blood. 2019;133(24):2575-85. doi: 10.1182/blood.2019 000478
- Imi T, Katagiri T, Hosomichi K, Zaimoku Y, Hoang Nguyen V, Nakagawa N, et al. Sustained clonal hematopoiesis by HLA-lacking hematooietic stem cells without driver mutations in aplastic anemia. Blood Adv. 2018;2(9):1000-12. doi: 10.1182/bloodadvances.2017013953
- Tichelli A, de Latour RP, Passweg J, Knol-Bout C, Socié G, Marsh J, et al. Long-term outcome of a randomized controlled study in patients with newly diagnosed severe aplastic anemia treated with antithymocyte globulin and cyclosporine, with or without granulocyte colony-stimulating factor: a Severe Aplastic Anemia Working Party Trial from the European Group of Blood and Marrow Trans- plantation. Haematologica. 2020;105(5):1223-31. doi: 10.3324/haematol.2019.222562
- Peffault de Latour R, Kulasekararaj A, Iacobelli S, Terwel SR, Cook R, Griffin M, et al. Eltrombopag Added to Immunosuppression in Severe Aplastic Anemia. N Engl J Med. 2022;386(1):11-23. doi: 10.1056/NEJMoa2109965
- Narita A, Muramatsu H, Ichikawa D, Hamada M, Nishikawa E, Suzuki K, et al. Relationship between plasma rabbit anti-thymocyte globulin concentration and immunosuppressive therapy response in patients with severe aplastic anemia. Eur J Haematol. 2021;107(2):255-64. doi: 10.1111/ejh.13644
- Zhang F, Zhang L, Jing L, Zhou K, Wang H, Peng G, et al. High-dose cyclophosphamide compared with antithymocyte globulin for treatment of acquired severe aplastic anemia. Exp Hematol. 2013;41(4):328-34. doi: 10.1016/j.exphem.2013.01.001
- LiL,LiY,LinL,YinJ,XuJ,WeiJ,etal.Outcomes of allogeneic haematopoietic stem cell transplantation for patients with severe aplastic anaemia using the porcine antilymphocyte globulin-containing conditioning regimen. Ann Hematol. 2020;99(8):1863-71. doi: 10.1007/s00277-020-04111-5
- Bernard F, Uppungunduri CRS, Meyer S, Cummins M, Patrick K, James B, et al. Excellent overall and chronic graft-versus-host-disease-free event-free survival in Fanconi anaemia patients undergoing matched related- and unrelated-donor bone marrow transplantation using alemtuzumab-Flu-Cy: the UK experience. Br J Haematol. 2021;193(4):804-13. doi: 10.1111/bjh.17418
- Lin F, Zhang Y, Han T, Cheng Y, Mo X, Wang J, et al. A modified conditioning regimen based on low-dose cyclophosphamide and fludarabine for haploidentical hematopoietic stem cell transplant in severe aplastic anemia patients at risk of severe cardiotoxicity. Clin Transplant. 2022;36(1):e14514. doi: 10.1111/ctr.14514
- Bradbury CA, Pell J, Hill Q, Bagot C, Cooper N, Ingram J, et al. Mycophenolate Mofetil for FirstLine Treatment of Immune Thrombocytopenia. N Engl J Med. 2021;385(10):885-95. doi: 10.1056/ NEJMoa2100596
- WuY,YanL,WangH,LiuH,XingL,FuR,etal. Clinical study on empirical and diagnostic-driven (pre-emptive) therapy of voriconazole in severe aplastic anaemia patients with invasive fungal disease after intensive immunosuppressive therapy. Eur J Clin Microbiol Infect Dis. 2021;40(5):949-54. doi: 10.1007/s10096-020-04054-9
- Jaiswal SR, Bhakuni P, Zaman S, Bansal S, Bharadwaj P, Bhargava S, et al. T cell costimulation blockade promotes transplantation tolerance in combination with sirolimus and post-transplantation cyclophosphamide for haploidentical transplantation in children with severe aplastic anemia. Transpl Immunol. 2017;43-44:54-9. doi: 10.1016/j.trim.2017.07.004
- Zhao X, Feng X, Wu Z, Winkler T, Desmond R, Olnes M, et al. Persistent elevation of plasma thrombopoietin levels after treatment in severe aplastic anemia. Exp Hematol. 2018;58:39-43. doi: 10.1016/j.exphem.2017.09.006
- Lee JW, Lee SE, Jung CW, Park S, Keta H, Park SK, et al. Romiplostim in patients with refractory aplastic anaemia previously treated with immunosuppressive therapy: a dose-finding and longterm treatment phase 2 trial. Lancet Haematol. 2019;6(11):e562-e572. doi: 10.1016/S2352-3026(19) 30153-X
- Ise M, Iizuka H, Kamoda Y, Hirao M, Kida M, Usuki K. Romiplostim is effective for eltrombopag-refractory aplastic anemia: results of a retrospective study. Int J Hematol. 2020;112(6):787-94. doi: 10.1007/s12185-020-02971-1
- Zhao LP, Sicre De Fontbrune F, Contejean A, AbrahamJ,TerriouL,ChabrotC,etal.Nationwide survey in France on the use of romiplostim in patients with refractory severe aplastic anemia. Bone Marrow Transplant. 2019;54(7):1161-3. doi: 10.1038/s41409-019-0452-1
- BentoL,BastidaJM,García-CadenasI,García-To- rres E, Rivera D, Bosch-Vilaseca A, et al. Thrombopoietin Receptor Agonists for Severe Thrombocytopenia after Allogeneic Stem Cell Transplantation: Experience of the Spanish Group of Hematopoietic Stem Cell Transplant. Biol Blood Marrow Transplant. 2019;25(9):1825-31. doi: 10.1016/j.bbmt.2019.05.023
- Jang JH, Tomiyama Y, Miyazaki K, Nagafuji K, Usuki K, Uoshima N, et al. Efficacy and safety of romiplostim in refractory aplastic anaemia: a Phase II/III, multicentre, open-label study [published correction appears in Br J Haematol. 2021 May;193(3):682]. Br J Haematol. 2021;192(1):190-9. doi: 10.1111/bjh.17190
- Imada K, Obara N, Iida H, Imajo K, Maeda T, Usuki K, et al. Eltrombopag in Combination with Rabbit Anti-thymocyte Globulin/Cyclosporine A in Immunosuppressive Therapy-naïve Patients with Aplastic Anemia in Japan. Intern Med. 2021;60(8):1159-68. doi: 10.2169/internalmedicine. 6063-20
- Lengline E, Drenou B, Peterlin P, Tournilhac O, Abraham J, Berceanu A, et al. Nationwide survey on the use of eltrombopag in patients with severe aplastic anemia: a report on behalf of the French Reference Center for Aplastic Anemia. Haematologica. 2018;103(2):212-20. doi: 10.3324/ haematol.2017.176339
- Kako S, Kanda Y, Onizuka M, Aotsuka N, Usuki K, Tachibana T, et al. Allogeneic hematopoietic stem cell transplantation for aplastic anemia with pre-transplant conditioning using fludarabine, reduced-dose cyclophosphamide, and low-dose thymoglobulin: A KSGCT prospective study. Am J Hematol. 2020;95(3):251-7. doi: 10.1002/ ajh.25693
- Ochi T, Onishi Y, Nasu K, Onodera K, Koba- yashi M, Ichikawa S, et al. Umbilical Cord Blood Transplantation Using Reduced-Intensity Conditioning without Antithymocyte Globulin in Adult Patients with Severe Aplastic Anemia. Biol Blood Marrow Transplant. 2019;25(2):e55-e59. doi: 10.1 016/j.bbmt.2018.09.039
- Pulsipher MA, Lehmann LE, Bertuch AA, Sasa G, Olson T, Nakano T, et al. A study assessing the feasibility of randomization of pediatric and young adult patients between matched unrelated donor bone marrow transplantation and immu- ne-suppressive therapy for newly diagnosed severe aplastic anemia: A joint pilot trial of the North American Pediatric Aplastic Anemia Consortium and the Pediatric Transplantation and Cellular Therapy Consortium. Pediatr Blood Cancer. 2020;67(10):e28444. doi: 10.1002/pbc.28444
