
Introducción
En 1892, Emil Theodore Kocher describe, por primera vez, la presencia de alteraciones musculares (aumento de la masa muscular, debilidad y lentitud de los movimientos) en pacientes con cretinismo. En 1934, Robert Debré y Georges Semelaigne comunicaron a la Sociedad de Pediatría de París esta alteración muscular, con el título de “hipertrofia muscular generalizada en niños”.1 Un año después (1935) dan a conocer la historia completa de dos casos y, a partir de entonces, fue designada como Síndrome de Kocher-Debré-Semelaigne (SKDS).2
Es una entidad poco frecuente, descrita dentro de los síndromes atípicos asociados al HC,3de moderado a grave, no tratado y de larga evolución, cuya característica principal es la presencia de pseudohipertrofia y debilidad muscular, con reversibilidad total del cuadro clínico con la implementación del tratamiento hormonal sustitutivo o de reemplazo con tiroxina.4
El HC tiene una incidencia de 1:3500 - 4500 nacidos vivos. Las disgenesias tiroideas producen casi el 85 % de estos casos y los defectos hereditarios en la biosíntesis de hormona tiroidea cerca del 10 a 15 % de los mismos.5
La deficiencia de HT es una inusual causa de miopatía adquirida, aunque la debilidad muscular puede ser la queja en un 40 % de los pacientes; de estos, menos del 10 % desarrollan pseudohipertrofia muscular,6incluyendo sus dos variantes:
- Síndrome de Kocher–Debré–Semelaigne de presentación en niños.
- Síndrome de Hoffmann (HS) de presentación en adultos.7
La incidencia real de la miopatía hipotiroidea es difícil de establecer con precisión debido a la diversidad de criterios diagnósticos utilizados en la literatura y a la ocasional asociación del hipotiroidismo con otras patologías neuromusculares, como neuropatía, miastenia gravis o miopatías inflamatorias, principalmente.8
Se desconoce la fisiopatología de la pseudohipertrofia muscular,9 pero se acepta que, como consecuencia de la hipofunción tiroidea prolongada, existan anormalidades oxidativas de glicosaminoglicanos, probablemente mediado por dos mecanismos: un desorden mitocondrial hormono dependiente, que afecta la provisión de energía al músculo, y el exceso de TSH, que estimularía la síntesis y el depósito de mucoproteínas (ácido hialurónico) en el músculo, con la consecuente degeneración y atrofia de las fibras musculares.10
La falta de la HT repercute en muchas funciones metabólicas del cuerpo, incluyendo el sistema músculo esquelético. Existe una disminución de recambio de proteínas y alteración en el metabolismo de carbohidratos que conduce a la acumulación de glucógeno en el músculo, mientras incrementa la cantidad de tejido conectivo y depósito de mucopolisacaridos, que mantiene la apariencia de hipertrofia muscular.11
Se ha postulado que existen incrementos en el tejido conectivo y en el tamaño y número de las fibras musculares; en casos severos existe atrofia de las fibras musculares tipo II asociadas con centralización e incremento del número de núcleos; además de un aumento en el depósito de glucógeno.12 A su vez, también se ha encontrado cierta relación con la consanguinidad de los padres.10
Caso clínico
Se presenta un paciente masculino, de raza blanca, procedencia rural, con diagnóstico de HC a los 32 días de nacido, con valor elevado de TSH > 96mU/L obtenida de cordón umbilical y TSH > 96 mU/L confirmatoria elevada de muestra del talón y muy bajos niveles de T4 en 11.8 nmol/L, que evidencian un HC severo. Se inicia el tratamiento sustitutivo con levotiroxina sódica.
El paciente acude a nuestros servicios a la edad cronológica (EC) de 13 meses, presentando inestabilidad en sus controles hormonales, sin lograr eutiroidismo en toda esta etapa de seguimiento. Como elementos referidos por su área de salud, se reportan antecedentes maternos de útero bífido, historia perinatal de nacimiento a término de 39,2 semanas, eutócico, con buen peso (3770 g), talla (52 cm) y circunferencia cefálica (34 cm), con un puntaje de Apgar de 9/9. Entre las manifestaciones postnatales se encuentran: la caída del cordón umbilical a los 9 días, ictero fisiológico prolongado, hipotonía muscular generalizada con una fontanela anterior amplia de 3,5 x 3 cm y fontanela posterior abierta al momento del diagnóstico, presentando signos ligeros de infiltración mixedematosa en la cara y el dorso de las extremidades.
La ausencia de los núcleos de osificación (epífisis distales del fémur y proximales de la tibia) en la radiografía de edad ósea (EO), realizado al diagnóstico, como signo evidente de hipotiroidismo fetal severo.
En el primer control realizado en el INEN se obtiene TSH 23 mU/L [0,3-4], T4 42 nmol/L [50-170], T4 libre 8 ng/dL [10-22] y T3 0,4 pg/mL [2,30-4,20], coincidiendo con una dosis de levotiroxina sódica subóptima para su edad, a razón de 4,5 μg/kg/día.
Al interrogatorio, la madre refiere que al personal de salud le llamaba la atención desde el nacimiento la contextura muscular aumentada del niño, que contrastaba con la debilidad incrementada de manera progresiva, además de la constipación intensa y preocupante.
Al examen físico encontramos como datos positivos: un niño apático, con discreto infiltrado mixedematoso periorbitario, gran dificultad de inicio de la marcha, con inestabilidad en la misma, estructuras musculares aumentadas, de aspecto hipertrófico de los músculos de las extremidades inferiores, rectos femorales, pantorrillas y glúteos, de intensidad moderada, menos marcado en extremidades superiores, bíceps, tríceps, deltoides e interescapulales con manifestaciones de debilidad e hipotonía muscular generalizada, de ligera a moderada.
Se constata retraso del lenguaje, con apenas escasos signos de vocalización, y Test cognitivo de Brunet- Lezine con puntaje intermedio, para un coeficiente de inteligencia (CI) normal de 88 puntos.
Desde el punto de vista pondoestatural, sin dificultades, con P: 11 Kg [50 pc], T: 75 cm [50 pc], CC: 45,5 cm [50 pc], P/T: [97 pc], con una EO=EC con presencia de núcleos de osificación del carpo, grande y ganchoso.
Ante estos elementos clínicos, se plantea una alta sospecha de SKDS y se indican estudios para confirmarlo. Se realizan ajustes inmediatos en la dosis de sustitución con levotiroxina, duplicando la misma y estableciendo un incremento a razón de 8,2 μg/kg/ día, en rango para su edad, y se implementa estimulación intensiva del neurodesarrollo y programa de fisioterapia.
Los resultados de laboratorio fueron normales (perfil hematológico, función hepática, renal, lípidos y lipoproteínas, iones, glucemia, insulinemia, 17-hidroxiprogesterona, anticuerpos tiroideos) a excepción de los valores elevados de la CPK 560 U/L [10-190] enzima que marca daño o alteración muscular.
El electrodiagnóstico se realizó a través de la electromiografía (EMG), que mostró un patrón miopático con actividad de inserción normal, amplitud de acción de los potenciales reducida y potenciales polifásicos normales. El estudio de conducción nerviosa (ECN) evidenciaba disminución de la velocidad de conducción para nervios motores.
El estudio inmunohistoquímico realizado a través de biopsia del músculo gastrocnemio derecho con microscopía electrónica de alta resolución, encontró cambios sutiles en la agregación mitocondrial y de glucógeno, con ligera dilatación del retículo sarcoplasmático de configuración membranosa y desgarro de algunos miofilamentos.
Ante la confirmación diagnóstica de SKDS, se evalúa la respuesta terapéutica luego de lograr la normalidad y estabilidad de la función tiroidea a las 6 semanas siguientes al ajuste en la dosis sustitutiva de tiroxina (TSH 1,6 mU/L [0,3-4], T4 122 nmol/L [50 – 170],
T4 libre 14 ng/dL [10-22] y T3 2,8 pg/mL [2,304,20]) y de la normalización de la CPK 120 U/L [10-190] con un seguimiento exhaustivo de todos los controles, con la frecuencia establecida; se identificó que en un periodo de 6 meses el niño consigue establecer la marcha y lograr un desarrollo psicomotor adecuado para su edad. A los 9 meses se aprecia una regresión parcial de los signos clínicos, con mejoría de la pseudohipertrofia en cuanto a su intensidad, modificando su patrón de signos moderados a ligeros, con recuperación del tono muscular.
En la reevaluación del área cognitiva, a los 20 meses, el Test de Brunet-Lezine muestra un coeficiente de desarrollo normal con 96 de puntaje.
A los 3 años se clasifica como un HC permanente con Ecografía tiroidea: ausencia de tejido tiroideo in situ y Gammagrafía tiroidea con Tc-99: ausencia de captación glandular del radiotrazador, que confirma en su etiología una Atireosis como defecto por disembriogenesis.
Su desarrollo y comportamiento escolar es similar al de los niños de su edad. A los 6 años mantiene signos ligeros de pseudohipertrofia de los músculos de la cintura escapular, bíceps, tríceps, rectos abdominales, femorales y glúteos, con buen tono muscular. El inicio puberal se reporta a los 10.8 años, con curso normal y un adecuado desarrollo pondoestatural, con buena correlación entre la edad cronológica y ósea. En la última evaluación, a los 12 años, se remarcaban los músculos con ligera hipertrofia a nivel de las extremidades superiores (deltoides y bíceps), los músculos dorsales y los glúteos, a pesar de mantener un excelente control de la enfermedad y buena calidad de vida relacionada con la salud. (Figura 1)
Discusión
El SKDS es un síndrome clínicamente bien definido, más prevalente en varones, en edades entre los 18 meses y 10 años,aunque se han descrito casos en edades más tempranas, incluso en el periodo neonatal, y aunque no se conoce su incidencia real, se estima que la hipertrofia muscular podría ser inferior al 10 % de los pacientes con un hipotiroidismo miopático.13 Se continúan reportando en países en los que no hay programas de pesquisa universal en recién nacidos,4y se puede asociar tanto a formas congénitas de hipotiroidismo (atireosis, defectos en la síntesis de enzimas) como adquiridas (autoinmune).6
Existe una amplia gama de síntomas y signos, fundamentalmente relacionados con el hipotiroidismo: letargia o insomnio, mixedema facial, macroglosia, aumento de las fontanelas, ictericia mucocutánea, estreñimiento, cambios del estado de ánimo, cabello grueso, retraso del crecimiento y pseudohipertrofia muscular, como signo imprescindible, que afecta preferentemente al tronco y las extremidades, ofreciendo un aspecto musculoso, paradójicamente asociado a debilidad muscular, que en ocasiones conlleva dificultad para sentarse y para el control de la posición de la cabeza.14
El cuadro clínico específico de la miopatía hipotiroidea abarca un espectro amplio de síntomas que discurren de forma aislada o en combinación: calambres musculares, debilidad muscular (generalmente proximal simétrica), rigidez y/o dolor muscular, intolerancia al ejercicio, mialgias difusas que incrementan la rigidez y el volumen muscular. Al examen físico el agrandamiento muscular es firme, resistente, “gomoso”, un poco más débiles y más hipotónicos, presentan características de contracción y relajación muscular lentas y la fase de relajación de los reflejos de estiramiento muscular suele estar prolongada, la debilidad no es una prominente característica.15
Se evidencia una hipertrofia generalizada, envolviendo particularmente los músculos de las extremidades, dando los niños una apariencia “atlética”, “niños hércules”. La pequeña proporción corporal del niño, el bajo contenido de grasa y el desarrollo de mixedema contribuyen con esta semblanza atlética o hercúlea.16 La pseudohipertrofia es más llamativa en los miembros, tronco y músculos faciales en algunos casos reportados.17
Se ha descrito también hepatomegalia, que resulta de infiltración grasa (del hígado) y es reversible con terapia.18 En casos más graves, refieren la aparición de manifestaciones orofaciales,10 rabdomiolisis,19cardiomiopatía arritmogénica del ventrículo derecho y derrame pericárdico,20pseudopubertad precoz,21 así como síndromes de coagulación intravascular diseminada.22
El diagnóstico de sospecha es fundamentalmente clínico y analítico, apoyado por la respuesta favorable al tratamiento con tiroxina.
Analíticamente, se acompaña de un descenso en el título de HT, con un aumento de la hormona tirotropa (TSH) y de las enzimas musculares. La elevación de los niveles de la enzima CPK es de grado leve, no en proporción vista en distrofias musculares, que son los más próximos, clínicamente diferenciables al síndrome, considerando el grupo de edad.12 La elevación de los niveles de CPK no se correlaciona con la gravedad de las manifestaciones clínicas, pero se ha encontrado correlación entre los niveles de CPK y los de TSH. Esta elevación de las enzimas musculares puede estar presente antes de que aparezcan las manifestaciones del hipotiroidismo.8Se ha visto también la elevación de CPK en pacientes con rabdomiolisis con Tiroiditis de Hashimoto y SKDS. 4, 19
El estudio inmunohistoquímico y ultraestructural realizados en biopsia muscular obtenida de un niño con SKDS, enumeran varias alteraciones calificadas como inespecíficas,23 cuyos cambios ultraestructurales vistos en la biopsia retornan a la normalidad con el tratamiento.14
En el microscopio electrónico se demuestra edema mitocondrial, fragmentación miofibrilar, aumento de depósitos de glucógeno, gránulos lipídicos y dilatación del retículo sarcoplasmático.11, 21La microscopía de luz muestra nucleación central, variación en el tamaño, estructura de fibras musculares y anillos espirales abortivos.6, 24
Potencialmente, puede ser engañoso el diagnóstico de un desorden primario muscular, pero por la evidencia bioquímica de hipotiroidismo debe hacer pensar en un SKDS.12 En cuanto al diagnóstico diferencial, debe establecerse con otras enfermedades que implican la presencia de debilidad muscular crónica (en los casos en los que aparezca la misma), fundamentalmente si el tratamiento con levotiroxina no mejora la sintomatología, tales como polimiositis, miastenia gravis, distrofia muscular congénita, mielomeningocele, esclerosis lateral amiotrófica.2, 14, 25 Mucopolisacaridosis, síndromes de crecimiento excesivo como Beckwit-Wiedemann, Simpson-Golabi-Behemer,3así como el síndrome de Van Wyk-Grumbach.26
Hay un limitado reporte de la descripción de regresión de la pseudohipertrofia muscular, entre otras características clínicas del seguimiento, tras el inicio de la terapia de reemplazo con tiroxina. Por su parte, Vishal, et al.4reportan una notable regresión después de 6 meses, excepto por la talla final que puede aún, ser corta.8, 12Se evidenció en una niña de 11 años, con hipotiroidismo autoinmune tras 3 meses de estabilidad del tratamiento, una mejoría tanto en la sintomatología como en el volumen del músculo de la pantorrilla.14
Un programa de fisioterapia puede ser beneficioso para conseguir suprimir la rigidez muscular y conseguir un pleno potencial de fuerza muscular.8, 12
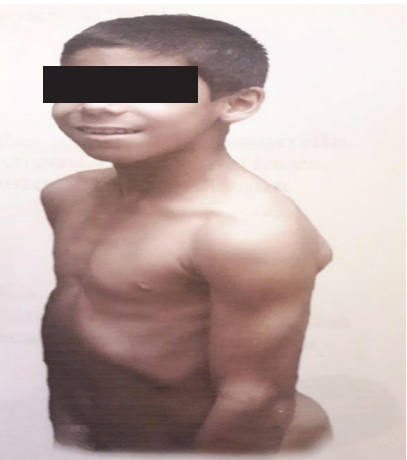
Figura 1. Apariencia del paciente con HC controlado y Síndrome de Kocher-Debré-Semelaigne a los 12 años de edad
Fuente: Instituto Nacional de Endocrinología.
Conclusiones
El SKDS constituye una de las formas atípicas de presentación del HC. Es rara su incidencia, sobre todo en la etapa neonatal, lo que no excluye su diagnóstico. La presencia de hipertrofia muscular debe considerarse un dato clínico de sospecha de hipotiroidismo, aun con la implementación de los programas de pesquisa neonatal. Es posible la regresión parcial de la pseudohipertrofia muscular con el restablecimiento de la función tiroidea en un periodo variable. El tratamiento con levotiroxina a las dosis establecidas es el de elección, con una respuesta muy favorable. Se debe tomar en cuenta en el diagnóstico diferencial de otras miopatías primarias, pues potencialmente puede ser engañoso el diagnóstico de estos desórdenes, pero por la evidencia de hipotiroidismo debe hacer pensar en un SKDS.
Conflicto de intereses
Los autores declaran no presentar conflicto de intereses que interfieran con la posible publicación del presente artículo.
