Ciencia y Salud, Vol. IV, No. 3, septiembre-diciembre, 2020 • ISSN (impreso): 2613-8816 • ISSN (en línea): 2613-8824 • Sitio web: https://revistas.intec.edu.do/
DÉFICIT DE HORMONA DE CRECIMIENTO EN NIÑOS Y ADOLESCENTES: ALGUNOS ASPECTOS A TENER EN CUENTA
Growth Hormone deficiency in children and adolescents: some aspects to consider
*Profesor y consultante de Endocrinología. Especialista de primer y segundo Grado en Endocrinología, Departamento de Endocrinología Pediátrica. Instituto Nacional de Endocrinología (INEN), La Habana, Cuba. ORCID: 0000-0002-8480-1360.
**Especialista de primer Grado de Endocrinología y en Medicina General Integral (INEN), La Habana, Cuba.
***Especialista de primer Grado de Endocrinología y en Medicina General Integral (INEN), La Habana, Cuba. ORCID: 0000-0002-4577-4486.
****Especialista en Bioestadística Médica, Profesor Auxiliar e Investigador Auxiliar. INEN, La Habana, Cuba. ORCID: 0000-0002-2289-0345.
*****Especialista en Medicina General Integral y de primer Grado de Oncología Clínica, Master en Enfermedades Infecciosas. Instituto Nacional de Oncología, La Habana, Cuba. ORCID: 0000-0002-1453-0526.
Recibido: ● Aprobado:

Cómo citar: Carvajal Martínez F, Bustamante Tejido M, Piz Ramos Y, Domínguez Alonso E, Carvajal Aballe M. Déficit de hormona de crecimiento en niños y adolescentes: algunos aspectos a tener en cuenta. cysa [Internet]. 18 de septiembre de 2020 [citado 18 de septiembre de 2020];4(3):63-70. Disponible en: https://revistas.intec.edu.do/index.php/cisa/article/view/1918
Introducción
Entre los trastornos del crecimiento, la baja talla constituye el motivo de preocupación fundamental en la consulta de endocrinología1,2 y, entre las causas endocrinas que pueden producirla, una de las más importantes es el DGH1,3-8. La deficiencia de hormona de crecimiento puede resultar de un daño en la producción hipotalámica de hormona estimulante de hormona de crecimiento (GHRH) o debido, a desordenes genéticos o congénitos que afectaron el desarrollo hipofisario. El DGH también puede ser secundario a lesiones del sistema nervioso central como tumores, cirugía, trauma, radiaciones o enfermedades inflamatorias o infiltrativas.9 Dado el número de causas genéticas de trastornos del crecimiento se está expandiendo rápidamente, estas condiciones también deberían ser excluidas. La rápida expansión del uso de paneles genéticos basados en el exoma, hace prever que, en un futuro próximo, un resultado negativo en un panel de causas genéticas conocidas pueda ser obligatorio10,11-16.
El déficit de GH puede ser hipofisario (primario), suprahipofisario (secundario), o bien, producirse por resistencia periférica a la GH o a los factores de crecimiento similares a la insulina (IGF) (periféricos).
El objetivo principal del tratamiento sustitutivo con hormona del crecimiento (GH) durante la infancia y la adolescencia es promover el crecimiento hasta alcanzar una talla adulta normal y, entre otros beneficios, lograr una maduración ósea adecuada. A partir de la introducción de la GH recombinante humana (rhGH) en 1985, miles de niños con una alteración del crecimiento se vieron beneficiados. La indicación principal para el tratamiento con rhGH sigue siendo la DGH en niños y adolescentes o adultos. Sin embargo, su uso se ha extendido a otras patologías que incluyen el síndrome de Turner, talla baja en niños que nacieron pequeños para su edad gestacional (PEG), el síndrome de Prader-Willi, la insuficiencia renal crónica, la talla baja por haploinsuficiencia del gen SHOX (short-stature homeobox-containing gene), la insuficiencia renal, en los últimos años se ha valorado también el uso de la hormona de crecimiento en la baja talla idiopática y en el Síndrome de Noonan.6,8,9,17-20
Una Guía Clínica desde la Sociedad de Endocrinología Pediátrica Americana se publicó en 201622, en ella se revisó, desde el punto de vista de la evidencia, la indicación de tratamiento en niños con DGH; en sus conclusiones se muestra contraria al uso rutinario de rhGH en todos los niños con talla inferior a -2,25 SDS. Concluye que la respuesta al mismo es muy heterogénea, con una ganancia media de talla de 5 cm, pero con una respuesta interindividual muy variable, por lo que considera que la decisión de tratamiento debe realizarse de forma individualizada, ya que muchos niños no responden al tratamiento. Recomienda que la decisión se tome tras explicar cuidadosamente a la familia riesgos y beneficios y que se evalúe cuidadosamente la respuesta al año, tanto desde el punto de vista físico como psicológico y de calidad de vida. En este estudio se sugiere individualizar la decisión de tratamiento en una toma de decisiones compartida con el niño y su familia, pero no establece criterios basados en la evidencia científica para tomar dicha decisión.
Con base en los estudios anteriormente mencionados, se plantea que la mejor respuesta se obtendría en los niños con padres más altos, mayor retraso en la edad ósea y mejor predicción de talla adulta al inicio del estudio, esto es, al grupo con probablemente mejor pronóstico de talla adulta sin tratamiento.3
El estudio de la maduración esquelética o edad ósea (EO) es un proceder rutinario de todos los departamentos de radiología pediátrica. Los pediatras y endocrinólogos reconocen este estudio como un reflejo de la maduración biológica de los niños23. En los niños con DGH, principalmente el grupo de los pacientes pre púberes, se encuentra inicialmente retrasada y muestra una aceleración tras el inicio del tratamiento sustitutivo con rhGH, el cual es más intenso durante la pubertad. Comúnmente en la práctica se monitoriza la evolución de la EO durante el tratamiento.23
En la actualidad, se puede afirmar de forma categórica que, en los niños con deficiente secreción de GH, el tratamiento con rhGH es efectivo, lo podemos evaluar con un seguimiento del grado de maduración ósea, entre otros factores19, 20. El inicio del tratamiento da lugar a periodos más o menos prolongados de intenso crecimiento, en relación con la gravedad y la duración de la deficiencia, en general, con una clara tendencia a alcanzar su propio percentil de crecimiento. Por tanto, deficiencias parciales o muy recientes ofrecen respuestas menos marcadas que las observadas en niños con deficiencias totales de GH y con una mayor evolución temporal18,21.
Se considera que la mejor y más simple guía para el diagnóstico del DGH es la realización y evaluación de la curva de crecimiento del paciente. Esto puede detectar un crecimiento lento desde los primeros meses de vida, aspecto que se incrementa en los años siguientes, hasta descender la talla por debajo del tercer percentil.
También se informa, como hemos señalado, que en niños con baja talla hipofisaria bajo tratamiento con GH, el mayor incremento en el crecimiento se logra en pacientes prepuberales18.
En el año 1992, realizamos un estudio donde se evalúa la respuesta al tratamiento con rhGH durante un periodo de dos años, en 17 pacientes (11 del sexo masculino y 6 del femenino), encontrando una mayor velocidad de crecimiento en pacientes del sexo masculino, y en quienes estaban comprendidos en edades prepuberales18. La respuesta al tratamiento fue mejor en los pacientes con déficit selectivo severo, en comparación con quienes tenían una deficiencia hormonal múltiple y en los que tenían un déficit parcial. A pesar de las diferencias encontradas en estos grupos se comprobó que el tratamiento con rhGH fue efectivo en todos los casos, y se produjo un incremento de mayor o menor magnitud de todas las dimensiones antropométricas evaluadas en este periodo, excepto en el pliegue cutáneo.
El tratamiento con hormona de crecimiento debe indicarse lo antes posible, igualmente cuando existe un hipopituitarismo con afectación de las esferas de TSH y/o de ACTH debe sustituirse de forma adecuada, posteriormente ya en edades alrededor de la pubertad, de existir déficit de gonadotrofinas, igualmente la esfera sexual deberá ser sustituida3,4,22.
Todo lo anteriormente expuesto, demuestra la importancia de este tipo de tratamiento y su inicio precoz en el manejo de los niños y adolescentes con baja talla por DGH, motivando la realización de este estudio con el objetivo de conocer algunos aspectos importantes a tener en cuenta durante el tratamiento con rhGH en los pacientes tributarios, con vista a mejorar la posible repercusión psicológica de estos, así como un mejor manejo y elevación de la calidad de la atención médica brindada a los mismos.
Método
Se realizó un estudio aplicado, retrospectivo y descriptivo de 108 pacientes seleccionados de un total de 190 historias clínicas de niños y adolescentes con el diagnóstico de baja talla hipofisaria por DGH, atendidos en el Departamento de Endocrinología Pediátrica del Instituto Nacional de Endocrinología (INEN), La Habana, Cuba. Del total de historias clínicas se escogieron aquellas (108) que contenían todos los datos necesarios para la investigación.
Método de obtención de los datos
Los datos relevantes de las historias clínicas se registraron en un modelo de recogida de datos adecuado. Se creó una base de datos automatizada en una hoja de cálculo electrónica Excel para su posterior procesamiento estadístico.
Criterios de inclusión y exclusión
Los criterios de inclusión fueron los siguientes:
- Ser niño o adolescente con un diagnóstico de baja talla hipofisaria por DGH (selectivo o asociado a otro déficit de hormonas hipofisarias) que haya sido confirmado en nuestro servicio.
- Tener una historia clínica con toda la información necesaria para nuestra investigación.
Se adoptaron los siguientes criterios de exclusión:
- Padecer de una enfermedad crónica no transmisible que interfiera con el tratamiento, tal como Asma Bronquial severa, nefropatía, cardiopatía, entre otras.
- Disponer de evidencias de incumplimiento del tratamiento con rhGH.
Ética de la investigación
La investigación fue aprobada por el Comité de Ética del INEN, se mantuvo en estricta confidencialidad los datos obtenidos. Los resultados solo se emplearán con fines científicos.
Resultados
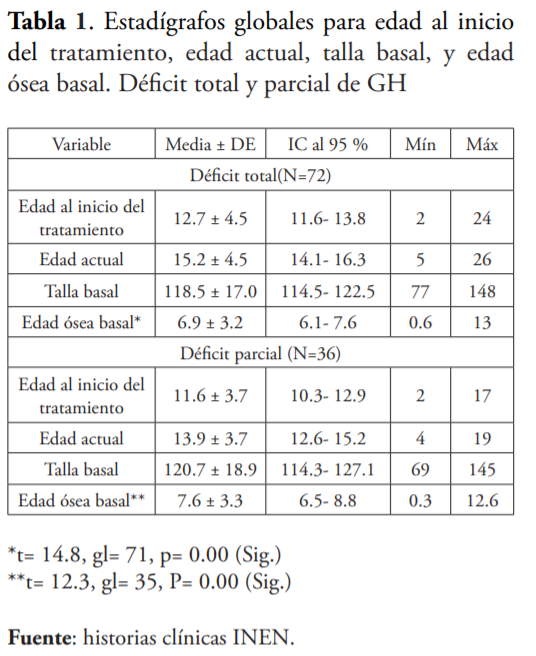
Se estudiaron un total de 108 niños de ambos sexos, 82 (75.93 %) niños y 26 (24.07 %) niñas. La edad de inicio del tratamiento es de 12.4 ± 4.3 años y la edad ósea basal (EOB) de 7.1 ± 3.2 años. La talla basal es de 120.4 ± 17.6 cm.
Cuando analizamos por sexo, observamos que en el sexo masculino (N=82) la edad al inicio del tratamiento es de 12.6 ± 4.3 años, la edad actual 15.1 ± 4.3 años, mientras la talla basal es de 120.4 ± 17.6 cm y la EOB de 7.1 ± 3.2 años. En el sexo femenino (N=26) la edad al inicio del tratamiento es de 11.5 ± 4.2 años, la edad actual 13.7 ± 3.8. La talla basal de 115.5 ± 17.4 cm y la EOB de 7.2 ± 3.3 años.
El déficit total representó el 66.67 % presentando una media de EOB (6.9 ± 3.2). Por otra parte, el déficit parcial (36,33.33 %), muestra una media de EOB (6.9 ± 3.2) hecho que se ilustra en la tabla 1.
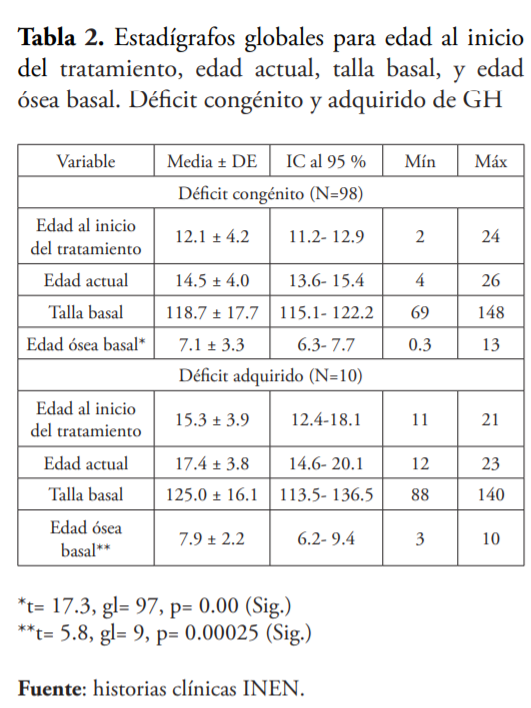
En la tabla 2, se analiza según la etiología del déficit. En el déficit congénito (98, 90.74 %) se inicia el tratamiento a la edad promedio de 12.1 ± 4.2 años, la talla basal es 118.7 ± 17.7 cm y presentan una EOB de 7.1 ± 3.3. Mientras el déficit adquirido (10, 9.26 %) se inicia tratamiento a los 15.3 ± 3.9 años, la talla basal es 125 ± 16.1 cm y presenta una EOB de 7.9 ± 2.2 años.
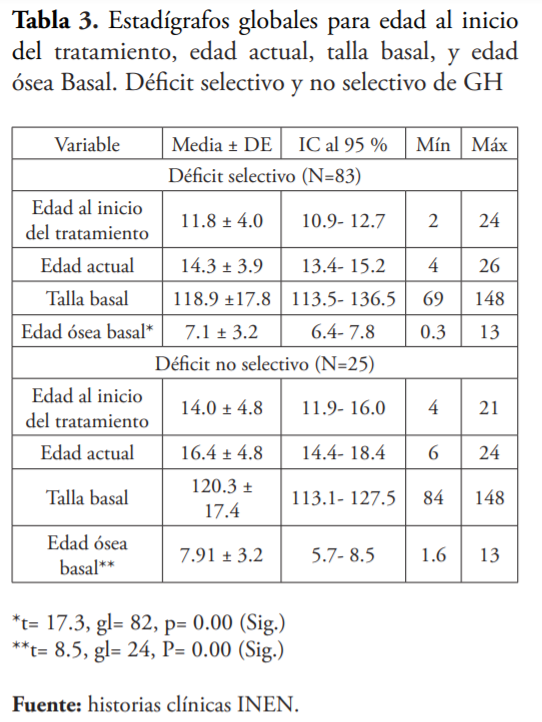
Las características, según la intensidad del DGH se analizan en la tabla 3, donde se constata que el déficit selectivo de GH inicia tratamiento a los 11.8 ± 4.0, una talla basal de 118,9 ± 17,8cm y una EOB de 7,1 ± 3,2 años. El déficit no selectivo inicia como promedio el tratamiento a los 14.0 ± 4.8 años, a la edad de 120.3 ± 17.4 cm una EOB de 7.91 ± 3.2 años.
Discusión
Debemos pensar en DGH cuando nuestro paciente tiene una baja talla armónica, o se encuentra en un percentil crítico, o ha tenido un descenso marcado percentilar, es decir, en su curva de crecimiento sin ninguna causa conocida. Frecuentemente, presentan manifestaciones clínicas clásicas: en los primeros dos años, antecedentes de hipoglucemia sin hiperinsulinismo, micro pene o criptorquidia, hipotermia en las primeras 24 horas o menor adaptación extrauterina del neonato, ictericia neonatal prolongada (más en los pacientes con déficit asociado de TSH), voz atiplada, así como, otras alteraciones genéticas, o de la línea media, entre otros aspectos.
Nuestro estudio muestra que la edad de inicio del tratamiento con rhGH tuvo una media de 12.4 ± 4.3 años, lo cual consideramos es un diagnóstico tardío dado las posibilidades de crecimiento para estos niños una vez iniciada la sustitución. Encontramos diferencias con otros estudios realizados9, 10, quienes iniciaron el tratamiento a una edad mucho menor. Consideramos que todavía debemos insistir, a nuestro nivel, en el diagnóstico de todo paciente con criterios posibles de DGH para lograr el inicio del tratamiento precozmente, aunque ya en la actualidad, tenemos pacientes con menos de 5 años que ya se encuentran bajo tratamiento con rhGH.
Se halló una diferencia estadísticamente significativa (p= 0.00) entre las medias de la edad de inicio de tratamiento y la edad ósea basal, la cual se encuentra muy retrasada en estos pacientes como resultado de la importancia que juega la GH y el factor de crecimiento similar a la insulina tipo I (IGF-1) en la maduración ósea, no existiendo diferencias entre sexos. Esta diferencia es más marcada en los niños que presentan déficit total de GH. El aspecto selectivo de la maduración ósea ha sido poco estudiado17, 23-26. En una investigación realizada por Rodríguez et al., donde se realizó un seguimiento de 30 niños por un periodo de tres años, los resultados obtenidos a través de las medidas de EO, se observó que estos niños inician el tratamiento con una relación EO/EC media menor de 1 y a lo largo del estudio se percibe una aceleración de la EO17. Según estos autores, no encontraron correlación entre el tratamiento con GH de forma general y cambios en la edad ósea, además constataron que el retardo de la edad ósea es menos intenso en las niñas.
En lo que respecta a la talla para la edad ósea, solamente el grupo de niños con deficiencia completa de GH muestra una mejoría significativa, ya que estos niños tienen muy retrasada la edad ósea inicial. En nuestra experiencia, en los pacientes con DGH tratados con hormona de crecimiento recombinante existió no solo incremento de talla, sino también de la maduración ósea.
Al parecer, la deficiencia aislada de GH es un poco más frecuente en los varones que en las hembras. Coincidimos con la mayoría de los estudios al encontrar un franco predominio del sexo masculino en pacientes con DGH, hecho descrito por la mayoría de la literatura que aborda este tema25. Diferimos en este aspecto del estudio realizado por Rodríguez et al., que informó un 60 % constituido por niñas17.
El vínculo entre la administración de la rhGH y la modificación de las características óseas en niños con déficit de esta hormona, ha sido objeto de atención para múltiples investigadores. En un estudio de 92 niños con déficit de hormona de crecimiento26, los autores observaron que estos niños han sufrido una disminución de la masa ósea antes del inicio del tratamiento y, en consecuencia, no logran alcanzar la máxima ganancia de esta masa, la cual ocurre en condiciones normales durante los primeros 4 o 5 años de vida y durante la adolescencia. Además, se concluye que la terapia de reemplazo con la rhGH proporciona la recuperación de la masa ósea de manera más pronunciada en el primer año de tratamiento, y previene la reducción progresiva manifestada en pacientes sin tratamiento anterior. Por consiguiente, el tratamiento con dicha hormona desempeña un papel importante en alcanzar la máxima ganancia de masa ósea en niños con el déficit en cuestión.
De acuerdo al origen del DGH, encontramos que cuando es congénito el diagnóstico es más precoz y la edad ósea está más retrasada que en las causas adquiridas, siendo estadísticamente significativo. Este hecho se justifica ya que previamente a la noxa que produjo el DGH el eje GH-IGF-1 funcionaba correctamente.
Podemos destacar, además, que cuando el DGH es selectivo la edad de inicio de tratamiento fue inferior, así como, la edad ósea estaba más afectada siendo estadísticamente significativo. También, es importante evaluar otros posibles factores que puedan retardar la maduración ósea, así como la importancia de procurar un tratamiento óptimo durante los dos primeros años de terapéutica. Opinamos, con relación a la maduración ósea, que se observa una disminución comparada con la edad cronológica, lógicamente mayor si la esfera tiroidea también está afectada. En nuestra investigación, el hecho que, al inicio del tratamiento la edad ósea basal fue significativamente inferior que la edad cronológica del sujeto en todos los grupos considerados, es indicativo de un marcado retraso de la edad ósea de nuestros pacientes.
Referencias
- Carvajal, F. Síndrome de Baja Talla. En: García R. Apuntes de Endocrinología Clínica. (2da ed.). Editorial Eclipse.Santo Domingo. 2016. p. 225.
- Gómez AL. Controversias en el tratamiento con hormona de crecimiento en la talla baja idiopática. Rev Esp Endocrinol Pediatr. 2018;9(1):59-63.
- Carvajal F, Pérez C. Síndrome de Baja Talla. En: Endocrinología Pediátrica. Libro de Pediatría. Cuba: Editorial Ciencias Médicas; 2011. Vol. VI: pp. 2510-2524.
- Carvajal F, González J, Güell R. Tratamiento de la baja talla hipofisaria con hormona de crecimiento. Informe preliminar. Rev Cub Ped. 1986;58:226.
- Carvajal F, Sánchez C, Güell R. Consideraciones terapéuticas sobre la absorción de hormona de crecimiento. Rev Cub Ped. 1987;59:463.
- Esquivel M, Richmont E. Diagnóstico y tratamiento del niño con déficit de hormona de crecimiento. En: Morla E. et al. Endocrinología Pediátrica Básica. Santo Domingo: Imp. Serigraf. 2019. pp. 29-42.
- Morla E. Estatura baja: Definición y Clasificación. En: Morla E. et al. Endocrinología Pediátrica Básica. Santo Domingo: Imp; 2019. pp. 19-21.
- Burgos M., Rivera L. Otras indicaciones aprobadas para el uso de la hormona de crecimiento. En: Morla E. et al. Endocrinología Pediátrica Básica. Santo Domingo Imp. Serigraf. 2019. pp. 43-48.
- Di Lorgi N, Morana G, Allegri AEM, Napoli F, Gastaldi R. Classical and non-classical causes of GH deficiency in the paediatric age. Best Pract Res Clin Endocrinol Metabol. 2016;30:705-36. Available from: http://dx.doi.org/10.1016/j.beem.2016.11.008
- Dauber A, Rosenfeld RG, Hirschhorn JN, Genetic evaluation of short stature. J Clin Endocrinol Metab. 2014; 99:3080-92.
- Wit JM, Oostdijk W, Losekoot M, van Duyvenvoorde HA, Ruivenkamp CA, Kant SG: Mechanisms in endocrinology: novel genetic causes of short stature. Eur J Endocrinol. 2016;174:145-73.
- Sergouniotis PI, Urquhart JE, Williams SG, et al. Agnathia-otocephaly complex and asymmetric velopharyngeal insufficiency due to an in-frame duplication in OTX2. J Hum Genet. 2015;60:199-202.
- Takagi M, Nagasaki K, Fujiwara I, et al. Heterozygous defects in PAX6 gene and congenital hypopituitarism. Eur J Endocrinol. 2015;172:37-45
- Xu H, Niu Y, Wang T, et al. Novel FGFR1 and KISS1R mutations in Chinese Kallmann syndrome males with cleft lip/ palate. Biomed Res Int. 2015; 1-9. Available from: http://dx.doi.org/10.1155/2015/649698.
- Williamson KA, Fitz Patrick. The genetic architecture of microphthalmia, anophthalmia and coloboma. Eur J Med Genet. 2014;57:369-80.
- Castinetti F, Reynaud R, Saveanu A, Jullien N. Mechanisms in endocrinology: an update in the genetic etiologies of combined pituitary hormone deficiency. Eur J Endocrinol. 2016;174:239-47.
- Rodríguez A, Rodríguez M, Laborta J, Martínez M, Alija M, Díez I, et al. Resultados tras el primer año de tratamiento con Hormona de Crecimiento Humana recombinante en un grupo de niños españoles con talla baja. Rev Esp Endocrinol Pediatr 2015;6(2):39-50.
- Romero, M. (1992). Experiencia en el tratamiento con hormona biosintética en niños con deficiencia idiopática de la hormona de crecimiento. (Tesis de Especialista de primer grado en Endocrinología). Instituto Nacional de Endocrinología. La Habana, Cuba.
- Saggese G, Baroncelli BL, Bertelloni S, Cinquanta L, Di Nero G. Effects of long term treatment with growth hormone deficiency. J. Pediatr 1993;122:37-45.
- Grimberg A, DiVall SA, Polychronakos C, Allen DB, Cohen LE, Quintos JB, et al. On behalf of the Drug and Therapeutics Committee and Ethics Committee of the Pediatric Endocrine Society. Guidelines for Growth Hormone and Insulin-Like Growth Factor-I Treatment in Children and Adolescents: Growth Hormone Deficiency, Idiopathic Short Stature, and Primary Insulin Like Growth Factor-I Deficiency. Horm Res Peadiatr. 2016;86:361-97.
- Wu, W. Cheng, X.D. Yang, B. Xiang. Growth hormone improves growth in pediatric renal transplant recipients-a systemic review and meta-analysis of randomized controlled trials. Pediatr Nephrol. 2013;28:129-33.
- Witz L, Josefsber Z, Kaufman H, Laron Z. When should hydrocortisone therapy be instituted in children with hypopituitarism? Am J Dis Child 1998;142:881-3
- Martin DD, Wit JM, Hochberg Z, Sӓvendahl L, van Ring RR, et al. The use of bone age in clinical practice. Horm Res Paediatr. 2011;76:1-9.
- Rodríguez S, Díez I, Sarasua A. Respuesta al tratamiento con hormona de crecimiento durante 3 años en niños: cambios en la composición corporal y somatometría. Bol. S VaSco-NaV PEdiatr. 2014;46: 5-11.
- Cancer Gaspar E, Ruiz Echarri Zelaya M, Labarta Aizpún JI, Mayayo Dehesa E, Fernández Longás A. Estudio de la masa ósea en pacientes con déficit de la hormona de crecimiento. An Pediatr. 2005;63(3): 219-23.
- Bajpai A, Kabra M, Grupta AK, Menon PS. Growth pattern and skeletal maturation following growth hormone therapy in growth hormone deficiencu: factors influencing outcome. Indian Pediatr. 2006;43(7):593-9.
