
Introducción
El síndrome de activación macrofágica (SAM) se caracteriza por la activación y proliferación benigna descontrolada de linfocitos T y macrófagos con sobreproducción de citoquinas, con fenómenos hemofagocíticos1,2. Se desarrolla como una alteración de los procesos de regulación inmune con diversas manifestaciones clínicas y datos de laboratorio como fiebre, hemofagocitosis de los histiocitos, hepatoesplenomegalia, citopenia, hipertrigliceridemia, hipofibrinogenemia, alteración de las enzimas hepáticas y coagulopatía3,4.
En la actualidad, este se incluye dentro de los síndromes hemofagocíticos secundarios, en la cual no hay afectación de las células de Langerhans. En diversas ocasiones no se asocian directamente con una enfermedad subyacente o factores desencadenantes, considerándose casos esporádicos.
Las manifestaciones cutáneas del SAM son paniculitis, edema y púrpura. La paniculitis histiocítica citofágica es el término de referencia de las manifestaciones cutáneas de esta patología; el hallazgo histopatológico es paniculitis lobulillar sin vasculitis con extensión a dermis profunda2,5.
La infección por el virus de Epstein-Barr se ha relacionado con linfomas cutáneos asociados a paniculitis citofágica histiocítica. Se ha sugerido que los casos benignos de paniculitis citofágica histiocítica son negativos para el virus de EpsteinBarr, y los asociados con linfoma, son positivos para el virus2.
Presentamos un caso de un varón con SAM, con paniculitis histiocítica citofágica de buen pronóstico, de origen esporádico.
Reporte de caso
Masculino de 13 años de edad quien presentó SAM con paniculitis histiocítica citofágica, sin antecedentes de importancia. Inicialmente, es evaluado en un centro de atención de tercer nivel por presentar fiebre no termometrada de 2 días de evolución, púrpuras y edema en miembros inferiores y superiores.
Se le diagnostica con absceso en cara interna del muslo derecho, es ingresado a sala, se transfunde y se maneja con antibioterapia desconocida y al no presentar mejoría, tras 15 días de tratamiento es referido a nuestro centro.
Al momento de su llegada, vía sala de emergencias, madre refiere historia de fiebre persistente de aproximadamente 18 días de evolución, la cual mejora con acetaminofén para luego reinstaurarse, lesiones purpúricas diseminadas y edema; por lo tanto, se decide su ingreso.
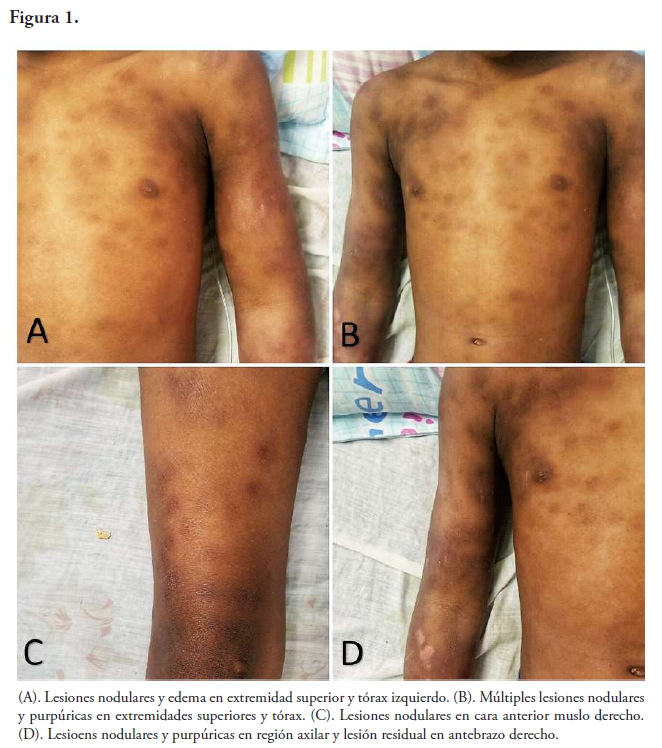
Al examen físico del paciente se encuentra consciente, alerta y orientado; presenta fiebre de 39° acompañada de edema en extremidades superiores, nódulos subcutáneos de 3-5cm de diámetro en extremidades superiores e inferiores, edema facial, púrpuras de bordes regulares en extremidades superiores e inferiores, y parte superior del tórax (región subclavicular y axilar anterior) de 3-4cm de diámetro.
Se realiza biometría hemática que muestra leucopenia (3400 leucocitos /mm3, GRAN 68.2 % y LINF 28.5%), anemia con (2.72 x 10^6/mm3), hemoglobina (6.9 g/dL), hematocrito (20.3%), VCM (75 pg), química sanguínea donde se observa hipoproteinemia (Albúmina 2.5 g/dL), hipertrigliceridemia (323 mg/dL), hipocolesterolemia 105 mg/dl, pruebas de función hepática que destacan ALT 142 U/L, AST 76 U/L, LDH 2094 U/L, prueba de proteína C reactiva (PCR) con resultado positivo; y perfil de coagulación que muestra TP 13.2-13.7s, TPT 26-28.8s y fibrinógeno 226 ng/ dL.
Durante su estancia en el centro de salud, se le realizan pruebas virales y se obtienen: IgG EBV (286 U/ml), citomegalovirus (102 U/ mL), IgG herpes simplex tipo 1 (índice 30.5) e IgG rubéola (80.2 UI/mL). HIV, HBsAg y HVC negativos.
Se realizan panel lúpico, factor reumatoide, Anti-TPO, C3, C4 y hemocultivo con resultado negativo.
El aspirado de medula ósea obtenido de la cresta ilíaca mostró hipocelularidad con megacariocitos disminuidos de 2-3/campo, linfocitos 21%, promielocito 8% y mielocito 18%; hemofagocitos 6% y otras células que asemejan células plasmáticas con resultado no concluyente de malignidad.
Se realiza biopsia de piel de nódulo subcutáneo localizado en muslo derecho, obteniendo un fragmento que mide 2 x 2 cms. La descripción microscópica evidencia intenso infiltrado inflamatorio a expensa de linfocitos e histiocitos a nivel de tejido celular subcutáneo y depósito de mucina intersticial superficial y profundo, así como el panículo adiposo, dando como diagnóstico histopatológico paniculitis lobulillar.
Inicialmente es tratado con terapia antibiótica por el departamento de infectología por considerarse proceso infeccioso. Al no mostrar mejoría, se inicia tratamiento con prednisona a dosis de 20 mg por día, durante 1 mes. Paciente evoluciona favorablemente y se suspende tratamiento con esteroides.
Discusión
El síndrome de activación macrofágica (SAM) es una entidad infrecuente y poco documentada debido, no sólo a la confusión generada por los diversos nombres que ha recibido en la literatura a lo largo de los años, sino también por su cercanía con otras patologías que cursan con un cuadro clínico muy similar, como lo es el síndrome hemofagocítico (SHF). La intervención y el manejo precoz son críticos, debido a falla multiorgánica rápidamente progresiva que resulta en morbilidad severa o mortalidad.
El SAM es una enfermedad de etiología incierta, aunque las infecciones virales, enfermedades sistémicas (como lupus eritematoso sistémico y artritis idiopática juvenil), toxicidad por aspirina y otros antiinflamatorios no esteroideos se consideran factores desencadenantes.
Su incidencia es desconocida a nivel mundial, esto se comprueba por la escasa cantidad de casos que figuran en la literatura2,6. Es típica en edad pediátrica, pero puede presentarse a cualquier edad.
Aparece por primera vez en la literatura con el nombre de linfohistiocitosis hemofagocítica, una enfermedad de la niñez2,7. Más tarde, en 1979 McKenna et al.1 la describen como un síndrome hemofagocítico relacionado a infección viral. Finalmente, es en el año 1973, cuando por primera vez se publica como SAM, relacionándolo a enfermedades inflamatorias sistémicas en niños. Actualmente se clasifica dentro de los síndromes hemofagocíticos secundarios o reactivos, asociados a enfermedades sistémicas, infecciones, inmunodeficiencias y malignidades6.
Algunos autores1,2,6–8 plantean que las manifestaciones clínicas del síndrome pudieran ser el resultado de alteraciones de los linfocitos T (estimulación de origen viral o neoplásica) y natural killer (NK), desencadenando una activación descontrolada de macrófagos por medio de la producción de mediadores inflamatorios; esto se evidencia por la presencia de hemofagocitosis, la cual es característica del síndrome y se considera una reacción celular benigna2,9,10or haemophagocytic lymphohistiocytosis (HLH.
El término hemofagocitosis describe la presencia de macrófagos activados que en su interior contienen linfocitos, eritrocitos, neutrófilos y/o plaquetas9,10or haemophagocytic lymphohistiocytosis (HLH. Esta excesiva activación de macrófagos o un defecto en la apoptosis, una vez activados, conllevaría a la instauración de una respuesta inflamatoria persistente con infiltración de los tejidos y producción de citoquinas, como el factor de necrosis tumoral (FNT) e interleukinas 1 y 6 (IL-1,IL-6), siendo éstas las principales responsables de la sintomatología2,7.
Recientemente ha sido descrita una mutación en el gen de la perforina (PRF1), la cual no estaría asociada a una alteración genética, sino más bien inducida por la enfermedad de base6. Dicha mutación causa una proliferación descontrolada de los linfocitos, que a su vez estimulan una producción excesiva de factor estimulante de colonia de granulocitos y macrófagos (GM-CSF)2,6.
Clínicamente se manifiesta de manera muy inespecífica, lo cual dificulta el diagnóstico temprano. En muchos casos puede aparecer con sintomatología indistinguible de una infección sistémica o artritis idiopática juvenil7.
El cuadro se caracteriza por la presencia de fiebre alta recurrente, linfadenopatías, hepatoesplenomegalia, disfunción del sistema nervioso central y hemorragias2,6,7. Las manifestaciones cutáneas más frecuentes son paniculitis y púrpura, las más específicas reciben el nombre de paniculitis histiocítica citofágica (PHC)10. Diversos reportes describen la PHC como lesiones nodulares, ulcerativas, costrosas y purpúricas en pacientes con síndrome hemofagocítico1,11,12. El hallazgo histopatológico típico comprende una paniculitis lobulillar con extensión a la dermis profunda, asociado a un prominente infiltrado linfohistiocitario2 .
Se ha sugerido que la PHC pudiera ser el resultado de un síndrome paraneoplásico o la respuesta a una infección por el virus Epstein Barr (VEB), el cual también se ha vinculado al linfoma de células T2,10.
En una serie de 10 casos presentada por Smith et al.10, 4 pacientes presentaron linfoma, de los cuales, 2 resultaron positivos para infección concurrente con el VEB. Asimismo, señalan que la infección viral no se excluye por la falta de pruebas positivas, ya que en muchos casos no se obtienen los estudios apropiados, o incluso, la inmunosupresión previa o una deficiencia inmune selectiva pueden no haber sido reconocidos. Partiendo de esta asociación, Marzano et al.13 establecen que la PHC pudiera ser un linfoma de células T de bajo grado, benigno, que en su fase más tardía, progresaría a una forma más agresiva denominada linfoma de células T paniculitis subcutánea, asociado a malignidad, destacando que ambas entidades representan la evolución natural de una misma patología.
La pruebas de laboratorio también son importantes para el diagnóstico del SAM, aunque no se presentan alteraciones específicas y ninguna de ellas es considerada gold standard2. Citopenia, elevación de enzimas hepáticas, lactato deshidrogenasa (LDH) e hipertrigliceridemia son algunos de los hallazgos más frecuentes, y usualmente, se acompañan de alteraciones en las pruebas de coagulación, hipofibrinogenemia y productos de degradación de fibrina como dímero-D2,6,9or haemophagocytic lymphohistiocytosis (HLH. No obstante, las enzimas hepáticas y pruebas de coagulación pueden estar normales al inicio14.
Recientemente, la elevación de la ferritina sérica se ha considerado un indicador de gran importancia (sensibilidad 90%, especificidad 96%) ya que puede alcanzar niveles muy elevados, por encima de los 10,000 ng/ml2,7,9de evolución fatal, secundaria a la activación del sistema fagocítico mononuclear, que se caracteriza por la proliferación incontrolada de los histiocitos con fenómeno de hemofagocitosis. Es un desorden de la regulación inmune caracterizado por fiebre, hemofagocitosis, hepatoesplenomegalia, pancitopenia, hipertrigliceridemia y coagulopatía. La etiología del SAM es desconocida pero se describen factores desencadenantes tales como infecciones virales y drogas. La excesiva activación y proliferación de los macrófagos bien diferenciados y de los linfocitos T induce una sobreproducción de citoquinas. Se ha postulado que existe una importante disminución de las células natural killer y un defecto funcional en la perforina. El curso clínico del SAM es variable. Clínicamente es similar a la linfohistiocitosis hemofagocitica reactiva asociada a virus y a la forma familiar. Las manifestaciones cutáneas más frecuentes son la paniculitis y la púrpura; la paniculitis histiocitica citofágica (PHC.
El hallazgo patognomónico del síndrome se localiza en la médula ósea, cuyo examen revela proliferación de histiocitos activados que fagocitan las células hematopoyéticas6. Estos hemofagocitos suelen no estar presentes en las etapas iniciales ni en todos los casos, por lo que se requieren de exámenes repetidos hasta que sea posible evidenciar dicha infiltración2.
El tratamiento inicial recomendado consiste en altas dosis de esteroides. En algunos casos este manejo puede no ser efectivo, y diversas guías recomiendan el uso de inmunosupresores y anticuerpos monoclonales9,14,15or haemophagocytic lymphohistiocytosis (HLH. En formas genéticas, el único tratamiento curativo es el trasplante de progenitores hematopoyéticos (TPH) para corregir el defecto de citotoxicidad9or haemophagocytic lymphohistiocytosis (HLH).
Conclusiones
La importancia de este caso radica en la baja tasa de diagnósticos bajo el título de SAM, debido a su confusión con linfohistiocitosis hemofagocítica o síndrome hemofagocítico, donde su diferencia reside en la manifestación cutánea del SAM llamada, paniculitis histiocítica citofágica, su incidencia sobre la población pediátrica y evidencia como casos aislados. El uso de terapia con corticoesteroides en dosis bajas es un punto por evaluar esta patología, debido a la buena respuesta del tratamiento en este caso. Asimismo, cabe destacar el impacto del diagnóstico y manejo precoz, disminuyendo considerablemente el riesgo de progresión a falla multiorgánica y la mortalidad por el síndrome.
Bibliografía
1. McKenna RW, Risdall RJ, Brunning RD. Virus associated hemophagocytic syndrome. Human Pathology. 1981 May;12(5):395-8.
2. Kouris E, Giansante E, Giansante Síndrome de activación macrofágica. Med Cutan Iber Lat Am. 2006;34(4):145-54.
3. Porras O. Síndrome de activación del macrófago. Revista Médica del Hospital Nacional de Niños Dr Carlos Sáenz Herrera. 2004;39(1):05-11.
4. Espinosa Bautista KA, Fossas PG, Eucario Y, Rodríguez L, Adriana K, Bautista E. Síndrome hemofagocítico. Conceptos actuales. Gaceta Médica de México. 2013;149:431-7
5. Astigarraga I, Fernández-Teijeiro A, García Pérez N, PiñánFrancés MA, Navajas Gutierrez A. Síndrome hemofagocítico en la infancia: A propósito de 4 casos. Anales Españoles de Pediatría. 1998;49(3):289-93.
6. Ravelli A. Macrophage activation syndrome. Current Opinion in Rheumatology. 2002 Sep;14(5):548-52.
7. Ravelli A, Minoia F, Davì S, Horne A, Bovis F, Pistorio A, et al. Classification Criteria for Macrophage Activation Syndrome Complicating Systemic Juvenile Idiopathic Arthritis: A European League Against Rheumatism/American College of Rheumatology/Paediatric Rheumatology International Trials Organisation Collaborat. Arthritis & Rheumatology. 2016 Mar;68(3):566-76.
8. McClain K, Eckstein O. Clinical features and diagnosis of hemophagocytic lymphohistiocytosis. [Internet] 2017 Jun [Cited Agos 2018] Available https://www.uptodate.com/contents/clinical-features-and-diagnosisof-hemophagocytic-lymphohistiocytosis
9. Astigarraga I, Gonzalez-Granado LI, Allende LM, Alsina L. Haemophagocytic syndromes: The importance of early diagnosis and treatment. Anales de Pediatria. 2018;89(2):124.e1-124.e8.
10. Smith KJ. Cutaneous Histopathologic, Immunohistochemical and Clinical Manifestations in Patients With Hemophagocytic Syndrome. Archives of Dermatology. 1992 Feb;128(2):193.
11. Crotty CP, Winkelmann RK. Cytophagic histiocytic panniculitis with fever, cytopenia, liver failure, and terminal hemorrhagic diathesis. Journal of the American Academy of Dermatology. 1981 Feb;4(2):181-94.
12. Cheah P-L, Looi L-M, Tan P-E, Bosco J, Kuperan P, Shi J, et al. Cytophagic histiocytic panniculitis. Chinese journal of pathology. 2005 Nov;34(1):59-60.
13. Marzano AV, Berti E, Paulli M, Caputo R. Cytophagic Histiocytic Panniculitis and Subcutaneous Panniculitis-like T-Cell Lymphoma. Archives of Dermatology. 2000 Jul;136(7):889-96.
14. Sawhney S, Woo P, Murray KJ. Macrophage activation syndrome: a potentially fatal complication of rheumatic disorders. Archives of disease in childhood. 2001 Nov;85(5):421–6.
15. Perotti A, Karakachoff M, Re AM, Pacitti M. Síndrome hemofagocítico asociado a infección aguda por virus de Epstein Barr. Arch Argent Pediatr. 2000;98(1):44.