
Introducción
La necidioblastosis se define como proliferación de las células endocrinas pancreáticas, que comporta alteraciones en su distribución y diferenciación. El número de los islotes de Langerhans es menor de lo normal, encontrándose las células endocrinas distribuidas, anárquicamente, por el parénquima pancreático, en forma de pequeños nidos o cordones1.
La hipoglucemia hiperinsulinémica endógena (HHE) puede ser causada por tumores sólidos del páncreas que secretan excesiva cantidad de insulina, conocidos como insulinomas, menos comúnmente por una hiperplasia de las células de los islotes de Langerhans, conocida como nesidioblatosis1, y rara vez por tumores secretores de factor de crecimiento insulinoide tipo 2 (IGF-2), tales como mielomas, linfomas y leucemias. La nesidioblastosis fue inicialmente reportada por George F. Laidlaw en 19382. La incidencia en niños se estima en 0.5 casos/año, sin diferencia entre sexos3.
Actualmente, gracias al trabajo de muchos investigadores, se han notificado 8 partes en el organismo, asociados al hiperinsulinismo: ABCC8, KCNJ11, HADH1, GCK, GLUD1, SLC16A1, UCP2 y HNF4a. Las mutaciones de estos sitios tienen diferencias importantes en el fenotipo y el modelo de herencia5.
Al respecto, los dos primeros, ABCC8 y genes de KCNJ11, se localizan juntos en el brazo corto del cromosoma 11, e involucra a los que presentan alteraciones del canal de potasio en la célula beta, lo cual la conduce a su despolarización con el ingreso de calcio y la consiguiente liberación de insulina. Por otra parte, el canal de potasio más adenosintrifosfato está formado por 2 pares de proteínas denominadas sulfonilureas (Sur-1 y Kir-6.2). Estas 4 subunidades forman un canal de potasio que se cierra en presencia de Atp. Dicha relación de defectos del receptor de sulfonilureas en la membrana de las células beta, con la consecuente alteración del flujo de potasio (ya mencionado) y el incremento de insulina no regulada por los mecanismos habituales de retroalimentación, da como resultado la hipoglucemia persistente5.
Caso clínico
Femenina de 7 años y 5 meses de edad, con historia de convulsiones tónico clónicas desde 1 año de edad, tratado con Oxcarbazepina; presentahipoglucemias sintomáticas al año y medio, por lo que su médico inició tratamiento con L- CARNITINA desde entonces, y hasta la fecha. A los 4 años se le suspenden anticonvulsivantes. Es referida desde otro centro de salud en Puerto Plata y tratada por endocrinólogo de adultos, luego de un internamiento por 9 días por hipoglucemias severas (181132-16 mg/dl…) y fiebre de 3 días de evolución, en condiciones generales muy grave.
Examen físico: sudoración profusa, sensación de mareos, temblor, estupor.
Tiroides: grado 0; Axilar: iii Olor apócrino, Mamas: tanner I; Púbico: tanner ii. Piel: suave. Patrón evacuatorio: diario.
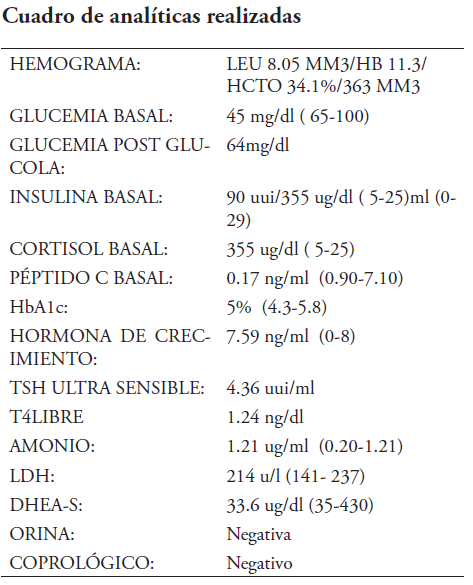
Peso: 33.7 kg (percentil 90-95). Talla: 123 cm (percentil 50). IMC: 22kg/m2 (percentil >97 correspondiendo a obesidad i) es trasladada al hospital infantil RRC, donde se estudia y se confirman las hipoglucemias, analíticas que revelan hormonas contrarreguladoras elevadas, niveles de insulina en 2,700 mui/ml en presencia de hipoglucemia 45 mg/dl.
Se realizan estudios por imágenes, como: sonografía abdominal, tomografía axial computarizada abdominal y resonancia magnética abdominal sin hallazgos patológicos.
Se corrige hipoglucemias con flujo de dextrosa 7mg/kg/min y octreótide vía parenteral, luego de estabilizar a paciente y dar de alta para continuar investigación del caso, se indica iazóxide jarabe para manejo ambulatorio. Sin embargo, este no lo administran, paciente vuelve a presentar hipoglucemias severas, acompañadas en dos ocasiones por eventos convulsivos. Se realizan analíticas y el resultado de Insulina basal es de 2,700 ui/ml, por lo que se decide optar por el tratamiento definitivo de las hipoglucemias secundarias a hiperinsulinismo: pancreatectomía para realizar biopsia por congelación. Se autoriza el procedimiento quirúrgico, y se realizan las evaluaciones prequirúrgicas correspondientes.
Informe quirúrgico: durante procedimiento se constata páncreas de gran tamaño por lo que se decide realizar Pancreatectomía Subtotal en un 95%.
Reporte de biopsia tranquirúrgica:
De tejido de cabeza de páncreas: tejido pancreático benigno con distorsión de los islotes de langerhans y presencia de células morfológicamente de tipo beta adyacente a los acinos serosos en el interior de los islotes bien delimitados. Estos hallazgos sugieren Nesidioblastosis. Sin embargo, se recomienda confirmar la presencia de células beta fuera de los islotes, por medio de inmunohistoquímica, ganglio pre arteria esplénica: ganglio linfático con hiperplasia linfoide reactiva benigna.
Paciente evoluciona satisfactoriamente y se egresa en condiciones generales estable, con tratamiento de:
• Análogos de insulina de acción lenta (0.14 ud/kg/d).
• Enzimas pancreáticas 10,000ud, 15 min previo a comidas.
• Multivitamínicos.
En las próximas dos semanas la paciente vuelve a presentar manifestaciones clínicas de hipoglicemia, mareos, temblor, pérdida de conocimiento y sudoración profusa, se suspende la insulina y al comprobarse las hipoglicemias se decide llevar nuevamente a quirófano y retirar el 5% restante de tejido pancreático. Actualmente en condiciones generales estables, la paciente no ha vuelto a presentar sintomatologías de hipoglucemia, reiniciando la insulina y continuando con enzimas pancreáticas y multivitamínicas.
Discusión
La hipoglicemia severa (por hiperinsulinismo endógeno) que amenaza la vida y que mejora momentáneamente con el aporte de glucosa, son características clínicas típicas del Insulinoma y la nesidioblastosis. Es difícil establecer la diferencia entre estas entidades desde el punto de vista clínico. Una vez que se ha excluido la enfermedad facticia del uso inapropiado de insulina o sulfonilurea7, el diagnóstico es eminentemente histológico. Para nesidioblastosis los criterios son:
a. aumento del tamaño y número de las células ß de los islotes de Langerhans, b. incremento del número de islotes periductales, c. núcleo hipercromático, d.-abundante citoplasma claro, e. exclusión microscópica e inmunohistoquímica de un insulinoma.2 De estas características las primeras dos coinciden con la histología de nuestra paciente, lo que confirma el diagnóstico de nesidioblastosis. Observamos en los estudios de imagen que con frecuencia no permiten evidenciar la lesión, siendo negativos los hallazgos en la nesidioblastosis, estudios como: sonografía abdominal, tomografía axial computarizada de abdomen y ecoendoscopia; se reportan sin hallazgos2, típico en la nesidioblastosis.
En el insulinoma, las pruebas de imágenes presentan una alta sensibilidad, principalmente en lesiones mayores de 2 cms, y localizadas en la cabeza y cuerpo del páncreas (TAC abdominal con contraste y multicortes con sensibilidad del 95%, ecoendoscopia sensibilidad 99%).2
Documentación de nivel de glucosa en sangre <50 mg / dl con síntomas de hipoglucemia; alivio de los síntomas después de comer, aumento del nivel de insulina en plasma (=6 µU / ml), aumento del nivel de péptido C (=0.2 nmol / l), aumento del nivel de proinsulina (=5 pmol / l), y ausencia de sulfonilurea plasmática.
Algunos autores sugieren un componente genético en la etiología de la Nesidioblastosis, como alteración en la expresión de los genes ABCC8 (SUR 1), KCNJ11 (Kir 6.2), GCK, GLUD1; en recién nacidos7, y en adultos, mutaciones secundarias a un patrón de herencia autosómico dominante, autosómico recesivo o a mutaciones de novo de los genes del SUR1 (gen ABCC8), o del canal recticador de potasio Kir 6.2 (gen KCNJ11)11.
En aquellos pacientes donde existe la sospecha clínica de un patrón heredo-familiar (al menos un familiar afectado en primer grado), se sugiere el estudio genético de consejería pre-concepcional para las próximas gestaciones; el estudio genético no está aún estandarizado para esta enfermedad, en el caso descrito no existen antecedentes familiares de sospecha para pensar en un componente heredofamiliar, por lo cual no fueron solicitados los estudios genéticos previamente señalados.
Conclusión
La nesidioblastosis es una causa rara de hipoglucemia hiperinsulinémica en la edad escolar. Los síntomas clínicos se basan en la presencia de la triada de Whipple (síntomas de hipoglucemia, concentraciones de glucemia venosa disminuida, desaparición de síntomas tras la normalización de la glucemia); y son indistinguibles del insulinoma. Los estudios de imágenes no aportan datos específicos, los análisis de laboratorio se basan en la documentación del hiperinsulinismo endógeno. El diagnóstico es histológico, con cambios en la morfología de las células ß dadas por la hiperplasia e hipertrofia de las mismas, núcleos hipercrómicos y citoplasma claro abundante. El tratamiento es quirúrgico, siendo recomendada la pancreatectomía casi total por su bajo riesgo de recidiva y la terapia de sustitución con enzimas pancráticas e insulina.
Referencias
1. Clínica Universidad de Navarra. Diccionario médico [Internet] Navarra, Clínica Universidad de Navarra, [citado 2018 mar]. Disponible en http://www.cun.es/diccionario-medico/terminos/Nesidioblastosis
2. De Jesús J, Fung L, García F, Núñez. Nesidioblastosis en adolescentes: A propósito de un caso. Rev. Venez. Endocrinol. Metab. [Internet]. 2015 Mar [citado 2018 Abr 17];13(1):4853. Disponible en: http://www.scielo.org.ve/scielo.php?script=sci_arttext&pid= S1690-31102015000100006&lng=es.
3. Antunes JD, Gefner ME, Lippe BM, Lanau BM. Hypoglycaemia in children: differentiating hyperinsulinaemic from nonhyperinsulinaemic causes. J Pediatr. 1990;116:990-994
4. Then C, Apostolopoulos Y, Seissler J, Lechner A. Refractory Idiopatic Non- Insulinoma Pacreatogenous Hypoglycemia in an adult: Case Report and Review of the literature. J Pancreas. 2013;14:264-268.
5. Zaldívar Ochoa JR, Rodríguez Carballo A, Quesada CM, Martínez AM, Santiago Oconnor A, Menéndez Rodríguez M. Nesidioblastosis: hipoglucemia hiperinsulínica persistente en un recién nacido. MEDISAN [Internet]. 2012 Dic [citado 2018 Abr 16];16(12):1948-1953. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S1029-30192012001200019&lng=es.
6. Shin, JJ, Gorden P, Libutti SK. Insulinoma: pathophysiology, localization and management. Future Oncology. 2010;6(2):229–237. Disponible en http://doi.org/10.2217/fon.09.165,
7. Thompson SM, Vella A, Thompson GB, et al. Selective Arterial Calcium Stimulation With Hepatic Venous Sampling Differentiates Insulinoma From Nesidioblastosis. The Journal of Clinical Endocrinology and Metabolism. 2015;100(11):4189-4197. doi:10.1210/ jc.2015-2404