
Introducción
Los TNEP son tumores de células pequeñas y redondas derivadas del tejido blando y pertenecen a la familia del sarcoma de Ewing, estos se originan en sitios intracraneales o periféricos del Sistema Nervioso Central, aunque algunas literaturas han reportado casos en pulmón y páncreas teniendo un curso clínico agresivo1-2. Estas neoplasias son clasificadas como grado IV, según la Organización mundial de la Salud (OMS)3.
Los TNEP comprenden del 2% al 3% de los tumores primarios del SNC en niños, ocurren generalmente en la primera década de la vida (su incidencia es máxima entre el nacimiento y los cinco años de edad). Tienen predilección por los lóbulos cerebrales, siendo los lóbulos frontal y temporal los más frecuentemente afectados. Guarda estrecha similitud histológica con el Meduloblastoma, pero la evolución clínica apresurada de esta patología nos permite descartarla como posible diagnóstico4.
El diagnóstico patológico de los tumores embrionarios se basa principalmente en características microscópicas histológicas e inmunohistológicas. No obstante, los estudios genéticos moleculares se emplean cada vez más para subclasificar los tumores embrionarios. En la actualidad, se utilizan estos hallazgos genéticos moleculares para estratificar el riesgo y planificar el tratamiento.
Presentación del caso
Paciente masculino de 2 meses de edad, procedente de Villa Mella, Santo Domingo, República Dominicana. Viene por consulta presentando aumento del perímetro craneal de 7 días de evolución y fiebre de dos días de aparición. Presenta un esquema de vacunación incompleto con dosis de BCG única y Hepatitis B1, dosis al segundo día de vida.
El lactante manejaba las medidas antropométricas dentro de los límites normales, a excepción del perímetro cefálico PC: 43.5 cm (percentil 97), siendo descrito como un lactante eutrófico macrocefálico. Al examen físico se evidencia fontanelas abombadas.
Se realizan analíticas de ingreso (Tabla I en anexos) y radiografía de tórax (Imagen I) que no exponen patologías aparentes. El paciente se ingresa con el diagnóstico de meningitis bacteriana.
Se recibe en sala un lactante alerta, hidratado, afebril, adecuada dinámica respiratoria, tolerando vía oral, en condiciones generales estables. Manejando unos signos vitales:
Valores de referencia
Ta: 80/50 mmHg Sistólica 50-75/
Diastólica 30-50 Fc: 152 l/m 120-180 l/m
Fr: 48 r/m 30-50 r/m
Temp: 37 0C
Se maneja con hidratación solución mixta 0.33%, 500 ml p/24h a 25ml/h endovenoso y medicado bajo ceftriaxona (80mg/kg/d) vancomicina (60mg/ kg/d) y acetaminofén (10mg/kg/d)
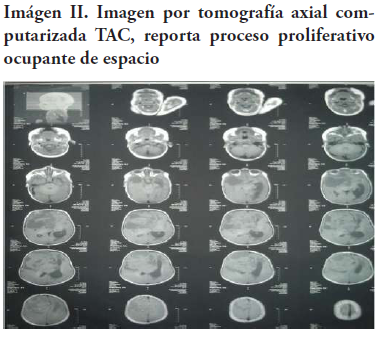
Al tercer día el paciente se torna quejumbroso, irritable y presenta cuadro febril termometrado en 40.5oC, por lo que se decide realizar imágenes que reportan un voluminoso proceso proliferativo con componentes quístico o necrótico que ocupa prácticamente la cuarta parte de la cavidad intracraneal en la región frontal del lado derecho (Imagen II).
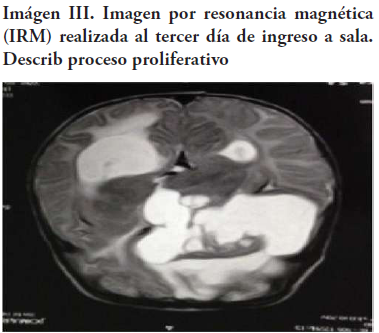
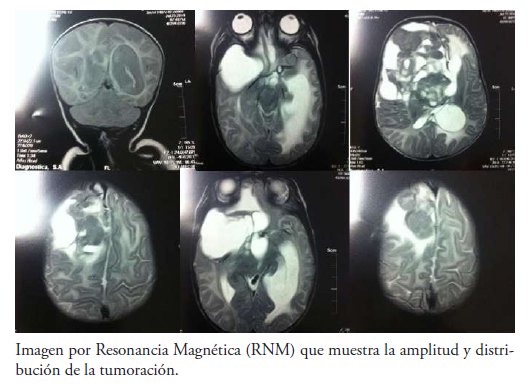
Se practicaron por interés científico secuencias en resonancia magnética que, en efecto, confirman las características de la lesión descrita en la tomografía. Se expone el diagnóstico de proceso proliferativo frontal derecho e hidrocefalia secundaria (Imagen III).
Para el sexto día de ingreso presenta un perímetro cefálico de 46 cm percentil 97, un estado irritable y se transfunde con paquete globular ORH(+) 10ml/ kg.
Al décimo día el paciente se presenta hipoactivo, quejumbroso y presenta signos meníngeos, como la rigidez de nuca. Presenta una eventualidad de evacuaciones líquidas, amarillentas, fétidas. Se realiza coprológico que reporta Entamoeba histolyca, y se medica con metronidazol.
Después de 24 horas, se decide trasladar a unidad de cuidado intensivo pediátrico (UCIP), el lactante se recibe hipertenso 110/70mmHg, por presentar movimientos tónico-clónicos generalizado con desviación de la mirada, relajación de esfínter y salida de abundantes secreciones a través de cavidad oral, así como exacerbación de dificultad respiratoria, anisocoria y períodos de apnea, por lo que se procede a manejar con tratamiento de diazepam e intubación endotraqueal, comunicando al servicio de neurocirugía, quienes consideran manejar con manitol. En UCIP se maneja con dieta cero vía oral, hidratándose con solución mixta 0.33% 500ml p/24h a 25ml/h endovenoso, 18 Meq Sol 24h. Medicado con Ceftriaxona (100mg/kg/d), Vancomicina(60mg/kg/d), Manitol (1g/kg/dosis C/6h), Albumina 1g/kg/d, Dexametasona 1mg/ kg/d, Difenilhidantoina 5mg/kg/d, Acetaminofen 10mg/kg/d, Keppra 30mg/kg (mantenimiento 30mg/kg/d), Midazolam infusión. 5mc /kg/min, Fentanilo 2/kg/h.
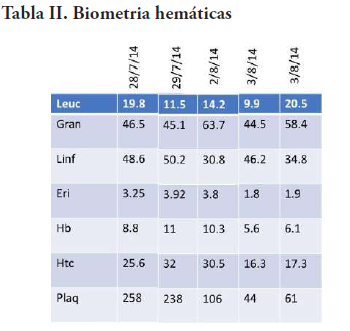
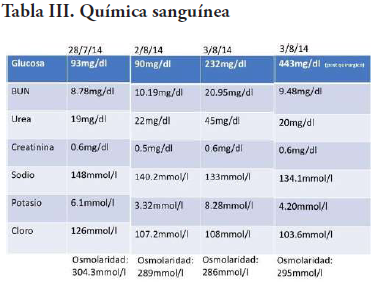
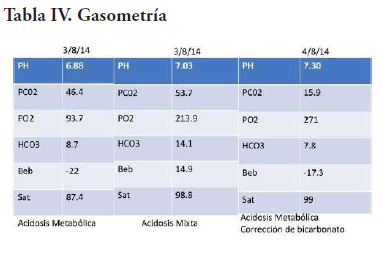
Al día siguiente, ya estable, se le realiza un procedimiento quirúrgico por parte del departamento de neurocirugía, donde se punciona el quiste colectándose unos 40ml de líquido de coloración ambarino. En la corticotomía frontal, se aprecia lesión tumoral sólida de consistencia elástica, moderadamente vascularizada, que cede a la succión, de coloración carne de pescado con bordes identificables, y se procede a extirpar gran parte de la lesión. Al presentar cambios cardiovasculares se decide suspender el procedimiento. El paciente presenta paro cardíaco que se logra revertir. A este se le realizan biometría hemática, química sanguínea y gasometría seriadas (Tabla II, III, IV).
El paciente fallece al doceavo día de ingreso, tras una parada cardiorrespiratoria que no pudo ser revertida. Los diagnósticos post-mortem fueron lesión tumoral intracraneal fronto parietal derecha, post quirúrgico de exceresis sub total de tumor, hidrocefalia secundaria y amebiasis intestinal.
Se realiza histopatología que expone tumor maligno indiferenciado de células pequeñas consistente con un PNET. Se realiza inmunohistoquímica que reporta muestra cerebral de un tumor neuroectodérmico primitivo (PNET). Los resultados positivos para los marcadores de la cresta neural, junto con los resultados positivos para CD99 y los hallazgos morfológicos son consistentes con esta impresión.
Discusión
Tratar a un paciente donde las patologías de índole infeccioso presentan alta frecuencia de aparición y aunada a una clínica sugestiva de la misma, en primera instancia se ingresa y aborda el paciente pensando en una meningitis bacteriana.
El cuadro se maneja bajo el diagnóstico antes descrito, al no evidenciarse mejoría significativa y al presentarse en su tercer día de ingreso, irritable, quejumbroso y con pico febril termometrado en 40.5oC, se realizan imágenes que arrojaron datos de masa ocupante de espacio, y con fines de conocer la dimensión del cuadro, se realiza procedimiento quirúrgico por parte de Neurocirugía, presentando los hallazgos y eventualidades ya descritas.
El avance agresivo y apresurado de esta patología impidió que se le brindara un abordaje terapéutico adecuado, la literatura describe el tratamiento en todos los grupos de edad, incluyendo resección quirúrgica lo más amplia posible, seguida de radiación craneoespinal y quimioterapia sistémica; sin embargo, los TNEP no pineales pueden ser resistentes a algunos regímenes de quimioterapia, por lo que actualmente debe reconsiderarse el esquema a utilizar e investigar otros esquemas alternativos5 y la recurrencia local, es el patrón de falla más común6.
El impacto pronóstico de las características patológicas y genéticas del TNEP no ha sido completamente investigado. Sin embargo, se ha visto que características anaplásicas y/o de células grandes, amplificación de MYCN o MYCC, polisomías de cromosomas 2 y 8; se asocian a disminución de la sobrevidaiv. Se ha demostrado que ciertos parámetros clínicos y biológicos pueden ser utilizados para pronosticar la sobrevida del paciente, tales como el alcance del SNC y la edad al momento del diagnóstico. Según el Instituto Nacional del Cáncer, los pacientes con TNEP que presentan diseminación de la enfermedad tienen probabilidad de sobrevida a 5 años, de 10-30%, siendo esta a su vez muy precaria. Con respecto a la edad, al momento del diagnóstico, si este tiene menos de 3 años se caracteriza por tener un desenlace desfavorable. Nuestro paciente cumple los parámetros clínicos explicados, por lo que se entiende que su sobrevida podía ser menor de 5 años y, aun así, se hizo el intento de prolongar la sobrevida, aunque no se obtuvo el resultado esperado.
Conclusión
Los tumores TNEP son patologías poco frecuentes, y por este patrón de aparición, son de difícil diagnóstico, a la vez, presentan una evolución rápida y agresiva. A pesar de afectar a una minoría de pacientes pediátricos, el pronóstico global para los casos de tumores de este tipo es pobre. No se ha establecido un tratamiento óptimo para este cuadro, la resección total del tumor, la radioterapia y la quimioterapia adyuvante son la forma más común de tratamiento empleado a nivel mundial.
Un estudio publicado en el 2016 por el Departamento de Pediatría del Hospital de Siriraj, en Thailandia, describe la presentación atípica con la que estos tumores suelen presentarse. En este estudio, donde se describen dos casos donde el estudio de imagen fue la herramienta de aproximación al diagnóstico, y no únicamente la clínica del pacientev.
Esta patología no se presenta únicamente en pacientes pediátricos, también se han descrito casos en adultos, como en el presentado por la revista de neuro-oftalmología del Centro Médico Zambrano Hellion-Tec Salud, Monterrey, México6.
En los casos en que esta patología logra descubrirse y tratarse a tiempo, una de las complicaciones más frecuente posterior al tratamiento es la recurrencia local, tanto en adultos como en pediátricos7, siendo uno de los abordajes a la cirugía de la recidiva con Gamma Knife8.

Referencias
1. Kliegman R, Nelson W. Nelson Tratado de pediatría. Barcelona: Elsevier; 2013.
2. Brat DJ, Parisi JE, Kleinschmidt DM, et al. A review of changes introduced by the World Health Organization (WHO) classification of tumors of the central nervous system, 4th edition. Arch Pathol Lab Med. 2008;132:993-1007.
3. Biswas S, Burke A, Cherian S, et al. Nonpineal supratentorial primitive neuro-ectodermal tumors (sPNET) in teenagers and young adults: Time to reconsider cisplatin based chemotherapy after cranio-spinal irradiation. Pediatric Blood Cancer. 2009;52(7):796-803.
4. Parker JP, Mortara RH, McCloskey JJ. Biologic behavior of the primitive neuroectodermal tumor: significant supratentorial childhood gliomas. Surg Neurol. 1975;4:383-388.
5. Phuakpet K, Larouche V, Hawkins C, et al. Rare Presentation of Supratentorial Primitive Neuroectodermal Tumors Mimicking Bifocal Germ Cell Tumors: 2 Case Reports. J. Pediatr. Hematol Oncol. 2016Mar;38(2):e67–70.
6. Espino BP, Angelina MD, Khan K, et al. Suprasellar Primitive Neuroectodermal Tumor in an Adult. Journal of Neuro-Ophthalmology. 2016 Sept;36(3):299–303.
7. Rachael A, Lester C, Brown LJ, et al. Clinical outcomes of children and adults with central nervous system primitive neuroectodermal tumor. Journal of Neuro-Oncology. 2014;120:371–379.
8. Se YB, Kim DG, Kim SK, et al. Gamma Knife Surgery for Recurrent or Residual Supratentorial Primitive Neuroectodermal Tumors. Stereotact Funct Neurosurg. 2016;94:273-278.