
Introducción
El rabdomiosarcoma infantil es el tumor maligno de tejido blando más frecuente. Representa aproximadamente 3.5 % de los casos de cáncer en niños de 0 a 14 años de edad. Según el Intergroup Rhabdomyosarcoma Study, la máxima incidencia del rabdomiosarcoma embrionario genital se registra en menores de catorce años, cuya ubicación típica es la vagina. La localización cervical, cuatro veces menos frecuente, es propia de adolescentes y adultas jóvenes10.
El rabdomiosarcoma fue descrito inicialmente por Webner, en 1854. Es un tumor complejo y de gran malignidad que se origina en las células del mesénquima embrionario con capacidad para diferenciarse en células musculares esqueléticas1.
Se han descrito 3 tipos histológicos de rabdomiosarcoma: embrionario (6 %), alveolar (20%) y pleomórfico (1%). El primero, que se subdivide en las variedades botrioides y fusocelular, es el más frecuente en la niñez. Representa alrededor del 6070% de los casos de rabdomiosarcoma infantiles y en ese grupo etario1,4.
La localización vaginal es rara, encontrándose frecuentemente en infantes femeninas menores de 5 años de edad (90% de los casos) y cerca de las dos terceras partes del total durante los primeros dos años de vida5.
La etiología de este sarcoma es desconocida. Es una neoplasia altamente maligna, de origen multifocal1,4, que crece como masas voluminosas, polipoides, que pueden llenar y sobresalir de la vagina, con aspecto de racimo de uvas (botrioide)6. Su consistencia es gelatinosa, friable y desprende fragmentos, produciendo hemorragias vaginales e infecciones secundarias6.
El pronóstico de esta enfermedad está determinado por variables como el tamaño tumoral, el órgano comprometido, la edad del paciente, el resultado quirúrgico (R0/R1) y la presencia de metástasis13. De esta manera, niños con tumores localizados en vía biliar, vagina y región de la cabeza y el cuello, menores de 5cm de diámetro y que no presentan metástasis ganglionares y/o viscerales tendrían mejores tasas de supervivencia5,6.
La evaluación médica multidisciplinaria precoz y oportuna permitirá siempre establecer un diagnóstico y tratamiento adecuados que mejoren el pronóstico de quienes padecen esta enfermedad1. Por este motivo es imprescindible que a las consultas médicas especializadas en Ginecología infanto juvenil sean remitidas todas aquellas niñas en las que sus familiares o el propio médico observen cualquier anormalidad en su aparato genital.
Esta entidad, aunque es poco frecuente, puede presentarse en nuestro medio. Es por esto que perseguimos con este trabajo realizar la revisión del tema y la presentación de un caso típico de Sarcoma botrioide de vagina.
Método
Se revisó la historia clínica de una paciente ingresada con el diagnóstico de Sarcoma botrioide de vagina en el año 2017.
Además, se confeccionó una ficha donde se recogieron los siguientes datos:
• Nombre de la paciente
• Edad, sexo, raza
• Motivo de ingreso
• Historia de la enfermedad actual
• Datos positivos al examen físico
• Resultados de los estudios complementarios
• Tratamiento indicado
Presentación del caso clínico
Paciente femenina de 13 años de edad, con antecedentes mórbidos negados. Conocida en la consulta de ginecología cuando acude referida desde otro centro de salud con reporte histopatológico de laparotomía exploratoria más biopsia por congelación de cuña de ambos ovarios por masa anéxial: para ovario derecho: tumor del estroma de los cordones sexuales, que incluye tumor de células de la granulosa tipo juvenil y tumor de células de Sertoly-Leydig; y en ovario izquierdo: quistes foliculares. Se realiza inmunohistoquímica que reporta tumor de células de Sertoly-Leydig. Trae reporte de resonancia magnética de pelvis, masa a nivel de vagina distal, que desplaza estructuras adyacentes sin invasión local probablemente neoformativa, ovario derecho con alteración de contorno y pobremente delimitado. Marcadores tumorales (CA-125, Alfafetoproteína, HE4) dentro de parámetros normales. Los estudios de laboratorio, la citometría hemática, la química sanguinea, las pruebas de funcionamiento hepático y los tiempos de coagulación fueron normales.
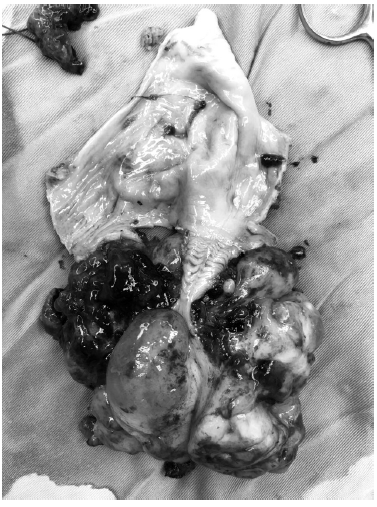
Con el diagnóstico ya mencionado se decide llevar a cirugía, donde se le realiza laparotomía exploratoria, toma de citología de líquido peritoneal, salpingooforectomía derecha, tumorectomía y omentectomía. En el transquirúrgico se evidencia una masa en la vagina que protruía a través del fondo de saco posterior de 10x12 cm., la cual se extirpa en su totalidad, no dejando lesión macroscópica visible.
El informe definitivo de patología mostró masa de vagina: sarcoma de bajo grado a favor de rabdomiosarcoma botrioide, tejido de ovario: tumor de células de Sertoly y Leydig de grado intermedio, omento: tejido fibroadiposo, citología de líquido peritoneal: negativo para malignidad.
Al mes de la cirugía, acude presentando dolor y distensión abdominal, por lo que se realiza TAC de abdomen y pelvis que reporta: gran masa pélvica, con componente sólido, con un componente superior que se adosa a la pared abdominal, pudiendo corresponder a implantes, sin que se descarte otra posibilidad, compresión de pared posterior de la vejiga y la pared anterior del recto.
Es llevada a cirugía nuevamente, donde se le realiza laparotomía exploratoria, evidenciando gran cantidad de tejido sarcomatoso. Se realiza citorreducción subóptima.
Al momento la paciente se encuentra en espera de inicio de protocolo de quimioterapia.
Estudios de extensión
Sonografia abdominal (9/03/2017): valoramos en cavidad abdominal imagen de masa heterogénea, que muestra en su interior áreas anecoides con relación a degeneración, diámetros aproximados de 12.8 x 8.0 cm y astitis.
Sonografía transvaginal (12/03/2017): lesión de masa anexial derecha, de 16.9 x 16.7 cm, líquido libre en cavidad pélvica.
Tomografía de abdomen (13/03/2017): lesión de masa compleja en cavidad abdomino-pélvica, a valorar lesión neoproliferativa, astitis, derramepleural bilateral.
Rx Tórax (16/03/2017): derrame pleural derecho.
Marcadores tumorales
Bhcg: 2 mU/ml, CA 125: 148 U/ml, Alfafetoproteína: 1.63 ng/ml
Discusión
Los rabdomiosarcomas representan el 50% de los sarcomas de tejidos blandos en menores de 15 años de edad; por tanto, es el sarcoma más común en la infancia con una incidencia de 4 a 7 niños por millón3,4. Corresponden al 5-8% de todos los tipos de cáncer en ese grupo etario12, con una prevalencia ligeramente mayor en el sexo masculino (relación 3:2)1. Alrededor del 50% de todos los rabdomiosarcomas ocurre en menores de 5 años de edad de ellos, solo un 2% está presente al momento del nacimiento1,3. La presentación cutánea primaria es extremadamente rara (0.7%)2,4 y, aunque escasas, las metástasis cutáneas pueden ser la primera manifestación de la enfermedad4. El tumor se origina en el mesénquima embrionario, precursor del músculo estriado fetal en las semanas 7 a 10,4,5; por tal razón, son tumores derivados de los rabdomioblastos o las células musculares primitivas. Esto se ha confirmado porque expresan los marcadores tumorales miogenina y mio D1 característicos de este tipo de neoplasias4. Los rabdomiosarcomas pueden aparecer en diferentes partes del cuerpo, obedeciendo a la siguiente distribución, por su frecuencia: cabeza y cuello, 35-40%; tracto genitourinario, 20%; extremidades, 15-20%; y tronco, pulmón o intraabdominal, 10-15%1.
La mayoría de los rabdomiosarcomas se desarrollan de forma espontánea, sin que exista algún factor predisponente conocido, pero en ocasiones ocurren en asociación con otros trastornos como la neurofibromatosis, el Síndrome de Costello, el Síndrome de Beckwith-Wiedemann, el Síndrome de Li Fraumeni, o en individuos con antecedentes de cáncer mamario materno4,6.
La variedad botrioides (del griego botrys, racimo; eidos, aspecto) es la forma polipoide del rabdomiosarcoma embrionario. Se caracteriza por múltiples proyecciones polipoides que forman racimos de consistencia gelatinosa, friables y que se desprenden en fragmentos, ocasionando hemorragias a menudo. Los tumores de este tipo representan cerca de un 10% de todos los casos de rabdomiosarcoma y suelen ser tumores embrionarios que se desarollan bajo la superficie mucosa de orificios corporales como la vagina y la nariz; también se ha visto afectado el tracto biliar3.
Dado su origen muscular, el diagnóstico puede apoyarse en la inmunohistoquímica positiva para actina, miogenina, desmina y vimentina3, mas el diagnóstico histopatológico diferencial debe contemplar toda la variedad de tumores malignos de células redondas como Sarcoma de Ewing, tumor neuroectodérmico primitivo y linfoma, en los que los marcadores musculares son negativos7. La presentación clínica del rabdomiosarcoma embrionario en la variedad botrioides es, en general, una masa que protruye por la uretra o el introito vaginal, o por la presencia de flujo fétido o sangrado vaginal en niñas menores de 2 años5,6. El pronóstico está determinado por variables como el tamaño del tumor, el sitio de origen, la edad del paciente, la enfermedad residual postquirúrgica y la presencia de metástasis al momento de establecer el diagnóstico.
Los sitios primarios de pronóstico más favorable incluyen órbita, paratesticulares, vagina y tracto biliar14. El tamaño del tumor al momento del diagnóstico tiene relevancia pronóstica. Los pacientes con neoplasias de tamaño inferior a 5cm tienen mejores expectativas de sobrevida que los individuos con tumores mayores de 5cm, en tanto que el pronóstico para los niños con enfermedad metastásica al momento del diagnóstico suele ser malo1.
La magnitud de la enfermedad residual posterior a la cirugía inicial también guarda correlación con el pronóstico. Según el Intergrupo de Estudio para Rabdomiosarcoma, la sobrevida a 5 años en pacientes con enfermedad residual voluminosa (Grupo Clínico III) es de, aproximadamente, un 70% contra 90% de sobrevida a 5 años en pacientes sin tumor residual postquirúrgico (Grupo clínico I) y un 80% de sobrevida a 5 años para pacientes con tumor residual microscópico después de la cirugía (Grupo clínico II)1. Se ha postulado que el 3-9% de los tumores botrioides reinciden transcurridos unos 5 años de haber remitido la enfermedad, reduciendo la tasa de supervivencia a un 64%. Las reincidencias suelen ocurrir, con mayor frecuencia (95%), dentro de los 3 primeros años posteriores al diagnóstico y al tratamiento, y son más frecuentes en pacientes con diagnóstico de rabdomiosarcoma estado III o IV2,5.
El tratamiento se fundamenta en el estadio clínico, la localización del tumor primario y la extensión de la enfermedad. Se centra en conseguir un “control local” y un “control sistémico”. El control local hace referencia a la erradicación permanente del “tumor primario”. Esto se lleva a cabo habitualmente mediante extirpación quirúrgica o irradiación del tumor (o ambos) y de cualquier área cercana afectada, añadiendo además tratamiento quimioterápico9,11.
Todos los pacientes con rabdomiosarcoma requieren quimioterapia para aumentar al máximo las posibilidades de curación. La mayoría de los niños en los Estados Unidos son tratados siguiendo los preceptos de un ensayo clínico internacional inicialmente denominado Estudio Intergrupo del Rabdomiosarcoma (ahora conocido como Comité de sarcomas de Partes Blandas del Grupo de Oncología Pediátrica).
Los fármacos más comunmente utilizados en el tratamiento del RMS en Estados Unidos y en Europa son: Vincristina, Dactinomicina, Ciclofosfamida, Topotecan, Irinotecan, Etopósido, Ifosfamida, Doxorrubicina y Carboplatino15.
Las niñas con rabdomiosarcoma embrionario del tracto genital (vagina, vulva, cérvix y útero) para las que la cirugía conservadora inicial es la regla pueden con frecuencia ser controladas mediante biopsias seriadas, comenzando aproximadamente 12 semanas después de completado el tratamiento quimioterápico, sin radioterapia adicional, aunque en casos seleccionados (generalmente restringidos a niños con RMS parameníngeo que ha erosionado la base craneal extendiéndose al interior de la bóveda), la radiación debe de comenzar al mismo tiempo que la quimioterapia (o tan cercana como sea posible).
Dependiendo de la localización, el tamaño y el grupo en que se incluya el tumor, se administran entre 20 y 28 sesiones de radioterapia8. De forma óptima, el tratamiento ha de planificarse sobre imágenes tridimensionales del tumor de forma previa a la toma de biopsia y al suministro de quimioterapia. Nunca será suficientemente enfatizada la importancia de la habilidad del oncólogo radioterapeuta en el éxito del tratamiento del RMS. Puesto que se trata de tumores poco frecuentes, y dado que la mayoría de los niños con RMS son tratados mediante protocolos en los que se especifica de forma detallada la terapia a seguir en cada caso, el oncólogo radioterapeuta debe de ser capaz no solo de interpretar con rigor los estudios de imagen relevantes para diseñar un “campo de tratamiento” apropiado, que abarque la totalidad del tumor original, añadiendo además un margen adicional de tejido circundante, sino también de llevarlo a cabo en el tiempo especificado en el protocolo, teniendo en cuenta la tolerancia del tejido normal en las estructuras sanas circundantes y los riesgos de complicaciones a largo plazo derivados de la irradiación de tejidos en crecimiento en niños pequeños.
Bibliografía
1. Monroy Prado GA, Toledo Bahena ME, Valencia Herrera A, Ramírez Cortés E, Mena Cedillos C. Rabdomiosarcoma genitourinario variedad botrioides: informe de un caso. Dermatología CMQ. 2013 [citado 25 abr 2016];11(3):208-212.
2. Junco Gelpi DA, Blanco Trujillo F, Montoya Cardero E, Junco Anaya DM, Anaya Correoso SM Rabdomiosarcoma pleomórfico del muslo. MEDISAN. 2015 [citado 25 abr 2016];19(2):252.
3. Tapia O. Rabdomiosarcoma Embrionario Uterino. Aspectos Morfológicos e Inmunohistoquímicos. Int J Morphol 2011; [citado 25 abr 2016]; 29(4) 29: 1126-1129.
4. Figueroa Carbajal JJ, Cárdenas Cardós R, Rivera Luna R, Castellanos Toledo A. Rabdomiosarcoma, experiencia de siete años en el Instituto Nacional de Pediatría. GAMO [Internet]. 2010 [citado 25 abr 2016];9(5):198-207.
5. Oscar Tapia E. Rabdomiosarcoma Embrionario Uterino. Aspectos Morfológicos e Inmunohistoquímicos. Int. J. Morphol [Internet]. 2011 [citado 25 abr 2016];29(4):1126-1129. Disponible en: http://www.scielo.cl/scielo.php?pid=S071795022011000400009&script=sci_arttext
6. Walterhouse DO, Meza JL, Breneman JC, Donaldson SS, Hayes-Jordan A et al. Local control and outcome in children with localized vaginal rhabdomyosarcoma: a report from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group. Pediatr Blood Cancer [Internet] 2011 [citado 25 abr 2016];57(1):76-83. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3459820/
7. Wharam MD, Meza J, Anderson J, et al. Failure pattern and factors predictive of local failure in rhabdomyosarcoma: A report of Group III patients on the Third Intergroup Rhabdomyosarcoma Study. Journal of Clinical Oncology 2004; 22 (4):1902-1908.
8. Schuck A, Mattke AC, Schmidt B, et al. Group II rhabdomyosarcoma and rhabdomyosarcomalike tumors: Is radiotherapy necessary? Journal of Clinical Oncology. 2004;22 (3):143-149.
9. Raney RB, Stoner JA, Walterhouse DO et al. Results of treatment of fifty-six patients with localized retroperitoneal and pelvic rhabdomyosarcoma: A report from the Intergroup Rhabdomyosarcoma Study-IV, 1991-1997. Pediatric Blood and Cancer. 2004; 42 (2):1-8.
10. Houghton JP, McCluggage WG. Embryonal rhabdomyosarcoma of the cervix with focal pleomorphic areas. J Clin Pathol. 2007;60 (5):88-9.
11. Maharaj NR, Nimako D, Hadley GP. Multimodal therapy for the initial management of genital embryonal rhabdomyosarcoma in childhood. Int J Gynecol Cancer. 2008;18 (2):190-2.
12. Labaran Dayyabu A, Adogu IO, Makama BS. Sarcoma botryoides a management dilemma: A review of two cases. Int J Case Rep Images [Internet]. 2014 [citado 25 abr 2016];5(7):482487. Disponible en: http://www.ijcasereportsandimages.com/archive/2014/007-2014-ijcri/CS-10044-07-2014dayyabu/ijcri-1004407201444-dayyabu-fulltext.php.
13. Pantoja Ludueña M, Riveros Moron A, Salvatierra Frontanilla I, Parra Nigañez P. Rabdomiosarcoma botrioide de vagina. Rev. bol. ped [Internet]. 2012 [citado 25 abr 2016]; 15(3): [Aprox.4p]. Disponible en: http://www.scielo.org.bo/scielo.php?script=sci_arttext&pid= S1024-06752012000300007.
14. Junco Gelpi DA, Blanco Trujillo F, Montoya Cardero E, Junco Anaya DM, Anaya Correoso SM. Rabdomiosarcoma pleomórfico del muslo. MEDISAN. 2015 [citado 25 abr 2016];19(2):252. Disponible en: http://scielo.sld.cu/scielo.php?pid=S102930192015000200014&script=sci_arttext&tlng=en.
15. Instituto Nacional del Cáncer. Disponible en: http://www.cancer.gov/espanol.