
Introducción
La craneosinostosis o craneoestenosis simple es una entidad patológica, de carácter idiopático, caracterizada por el cierre prematuro de las suturas de la bóveda craneana, dando como resultado una notable deformación, en la región de la sutura comprometida, secundada por una alteración en el crecimiento del cráneo.
Las craneosinostosis, de acuerdo a su grado de complejidad, pueden dividirse en: craneosinostosis idiopática o simples (escafocefalia, plagiocefalia anterior y posterior, trigonocefalia y braquicefalia), craneosinostosis sindromáticas (síndromes de Crouzon, Apert, Chotzen, Pfeiffer y de Carpenter), craneosinostosis secundarias, craneosinostosis relacionadas con enfermedades hematológicas, metabólicas y teratógenas.
El Síndrome de Crouzon o dismorfia cráneo facial, se conoce desde la antigüedad. Homero, el poeta griego, en su obra clásica La Ilíada, describe un guerrero llamado Tersites: “el hombre más feo fue el que vino de Troya”, “su estrecha cabeza”; esto se conoce como una de las primeras alusiones a las deformidades craneales tipo craneosinostosis. En 1791, se logra un paso de avance al plantear que el crecimiento del cráneo ocurre a lo largo de las suturas del calvario y que el fallo en su crecimiento provoca una enfermedad craneal.
A pesar de las investigaciones anteriores, no fue hasta 1851 en que Virchow inicia la verdadera etapa científica con un estudio anatómico completo.4 Pero, en 1912 es que describe por primera vez Crouzon al evaluar a una madre y su hija afectadas por una craneosinostosis que abarcaba diversas suturas, provocando una deformidad cráneo-facial junto a exoftalmos. Posteriores publicaciones médicas de casos similares configurarían este síndrome, recibiendo el nombre del primer autor.
La genética de este síndrome es compleja. Su herencia es autosómica dominante (AD), con penetrancia completa y expresividad variable.6 Dicho síndrome está producido por mutaciones del gen del receptor del factor de crecimiento de fibroblastos tipo 2 (FGFR2), que está localizado en la región 10q26. Además, se ha señalado también una entidad compuesta por la asociación del Síndrome de Crouzon con acantosis nigricans, planteándose que es el resultado de una mutación específica (ala391 a Glu),5,7 es decir, una mutación recurrente G1172A6 en el dominio transmembrana de otra proteína de la misma familia en el gen del receptor del factor de crecimiento de fibroblastos tipo 3 (FGFR3).
Solamente hay 35 casos publicados hasta la fecha,8 los cuales tienen un amplio espectro clínico por probable existencia de genes modificadores.
El riesgo de repetición es del 50% para los hijos de los afectados. Este síndrome presenta una gran variabilidad de expresión, por lo que es aconsejable asegurarse de que los padres de un afectado no presenten pequeñas manifestaciones. Existe una familia en la que se documentó la existencia de mosaicismo gonadal. Dependiendo de la presencia o ausencia del síndrome en alguno de los padres, el riesgo de repetición en hermanos puede variar entre un 50% y un riesgo muy bajo (el correspondiente a la existencia de un mosaicismo gonadal en alguno de los progenitores).
La frecuencia de aparición del síndrome en la población general oscila entre 1/25.000 y 1/60.000 nacimientos. Alrededor de la mitad de los casos son mutaciones nuevas (padres sanos). Como en otros síndromes de etiología AD, existe una clara relación de estos casos de mutaciones de novo con una edad paterna avanzada. Constituye el 4,8 % de todas las craneosinostosis y no tiene predominio racial ni de sexo.
El cuadro clínico es poco intenso al momento del nacimiento en la mayoría de los pacientes, pero a partir de los primeros meses de vida se encuentra dominado por la forma del cráneo y, de manera secundaria, por las alteraciones mentales, visuales y auditivas.
Los afectados por este síndrome pueden presentar variadas manifestaciones clínicas, entre ellas: craneosinostosis (generalmente de las suturas coronal, sagital y lambdoidea), exoftalmos (órbitas poco profundas), hipoplasia maxilar, braquicefalia, estrabismo divergente, anomalías del nervio óptico, abombamiento frontal, hipertelorismo, nariz en pico de loro, desviación del tabique nasal, labio superior corto, abombamiento lateral de las valvas palatinas, dientes apelotonados, erupción ectópica de los molares maxilares, atresia de conducto auditivo, calcificación del ligamiento estilohideo, fusión de vértebras cervicales (sobre todo C2-C3), retraso mental, convulsiones, agenesia de cuerpo calloso, hidrocefalia, cráneo en trébol, atrofia óptica, aniridia, anisocoria, nistagmus, coloboma de iris, microcórnea, megalocórnea, queratoconus, catarata, ectopia lentis, escleróticas azules, glaucoma, labio leporino, paladar hendido, úvula bífida, subluxación de la cabeza del radio, cúbito valgo, ectrodactilia, anomalías costales, anomalías sacras.12-15 Por el interés clínico que despierta y lo infrecuente de su aparición, asociada a una baja talla debido a una deficiencia de hormona del crecimiento (GH), consideramos importante presentar el caso de este paciente.
Presentación del caso clínico
Se trata de un paciente masculino de 12,9 años de edad cronológica, de piel blanca, procedente de La Habana, con antecedentes patológicos personales de Síndrome de Crouzon. Asiste a consulta de Endocrinología del Departamento de Endocrinología Pediátrica del Instituto Nacional de Endocrinología por presentar retraso del crecimiento y del desarrollo.
El paciente es producto de un embarazo de madre y padre con 16 y 25 años, respectivamente. Requirió un ingreso por bajo peso materno durante el segundo trimestre. Nació por un parto eutócico a las 39.1 semanas de edad gestacional, con un peso al nacer de 6.11 libras. Se desconocen otros datos perinatales. A los 6 meses de edad la familia comienza a notar el gran tamaño de los ojos. Este siguió incrementándose y a los 2 años de edad se interconsulta con especialidades afines, obteniendo el diagnóstico de Síndrome de Crouzon; en ese momento, se da a conocer que su padre y abuelo paterno padecían de la misma enfermedad.
En ese mismo año fue operado de craneosinostosis sin complicaciones. A los 5 años de edad sufrió un traumatismo craneal y fue operado de una fractura del cráneo donde le colocaron una prótesis frontal. Su crecimiento y desarrollo transcurrió normalmente, pero alrededor de los 12 años de edad cronológica sus padres comienzaron a notar diferencias de tamaño con respecto a los niños de su misma edad, es decir, notaban que estaba muy pequeño para su edad, motivo por el cual fue llevado a consulta en el Departamento de Endocrinología del Hospital Pediátrico Docente Juan Manuel Márquez.
En la primera consulta realizada en ese centro, se constató que su talla era de 127 cm y peso de 26 kg, con percentiles de peso y talla para la edad y sexo inferiores al tercero correspondiente para 8.6 años, la talla y 9.5 años el peso. Además, era examen físico se evidenciaron las manifestaciones clínicas características de su enfermedad de base, no se detectaron alteraciones a nivel de la glándula tiroides, ni de las paratiroides, sin presencia de vello sexual, con un pene en estadio Tanner I y ambos testículos de ± 6ml cada uno y en bolsas escrotales.
Posteriormente, se le realizaron exámenes complementarios que revelaron los siguientes resultados:
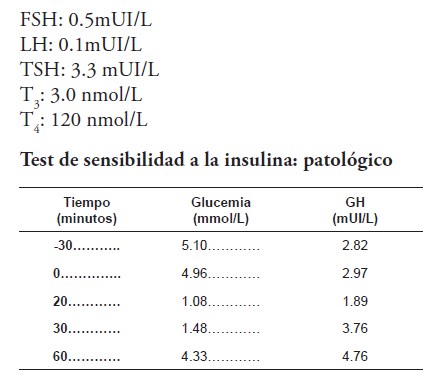
Radiografía de edad ósea: edad ósea aproximadamente 10 años (± 3 años de retraso de la edad ósea con respecto a la edad cronológica). Resonancia magnética nuclear de hipófisis: con cortes simples a 2 mm, sin observarse alteraciones intraselares.
Debido a estos resultados, fue remitido al Centro Nacional de Referencia ubicado en el Departamento de Endocrinología Pediátrica del Instituto Nacional de Endocrinología, donde se le diagnosticó finalmente una baja talla por deficiencia de hormona de crecimiento selectiva e idiopática asociada al Síndrome de Crouzon. Se indicó tratamiento con hormona del crecimiento a dosis de 1 mg/m2/día por vía subcutánea a las 9:00 pm, a los 14.5 años de edad cronológica, con una talla en aquel momento de 134.5 cm (talla para la edad por debajo del tercer percentil para su edad y sexo).
Ha mantenido un seguimiento periódico en consulta, teniendo como resultado que en un período de 14 meses ha crecido 11.2 cm. Ha alcanzado un estadío Tanner III- IV, con un pene de ± 12 cm de longitud y dividir de otro modo en bolsas escrotales con volumen de ± 12 ml cada uno, de consistencia fibroelástica.
Discusión
Los trastornos genéticos de mayor prevalencia asociados a craneosinostosis son: Síndrome de Apert o acrocefalosindactilia tipo I, Síndrome de Crouzon o acrocefalosindactilia tipo II, Síndrome de Carpenter o acrocefalopolisindactilia, Síndrome de Chotzen y Síndrome de Pfeiffer.
En la familia de las alteraciones del gen FGFR3, se encuentran agrupadas unas cuantas patologías, tales como acondroplasia, hipocondroplasia, displasia tanatofóricas tipo I y II, el Síndrome de Crouzon con acantosis nigricans, el Síndrome SADDAN, el Síndrome de Muenke y otros fenotipos asociados.
Las mutaciones en el mismo gen FGFR pueden dar lugar a diferentes síndromes de craneosinostosis, por lo que se plantea un mismo mecanismo patológico en el gen FGFR causante del Síndrome de Pfeiffer y el Síndrome de Apert. Estos últimos presentan algunas similitudes, pero con un buen examen físico se logra definir cada uno, y genéticamente también. El síndrome de Crouzon es fenotípicamente similar al de Pfeiffer, pero carece de las anomalías de pies y manos. A veces el Síndrome de Pfeiffer ha sido confundido con el de Saethre Chotzen-Jackson, por las características en los dedos de los pies, muy similares en ambos.
Por su parte, el Síndrome de Carpenter, con carácter autosómico recesivo, tiene múltiple fusión de suturas, cráneo con deformidad en hoja de trébol y retraso mental frecuente, entre otras anomalías.16 El síndrome de Chotzen es el de mayor prevalencia y se caracteriza por una craneosinostosis simétrica, plagiocefalia y sindactilia de tejidos blandos de los dedos dos y tres de las manos.
El síndrome de Pfeiffer presenta una herencia autosómica dominante y puede estar causado por dos tipos de mutaciones: la que afecta al gen FGFR1, situado en el cromosoma 8, o la del gen FGFR2, en el cromosoma 10.21 Se asocia a turricefalia, sinostosis radio humerales o radio cubitales, de ojos prominentes y muy separados, sus pulgares y dedos son gordos, cortos y anchos.16 Ocasionalmente, puede presentarse hidrocefalia, junto con protrusión ocular marcada, codos anquilosados y retraso del desarrollo psicomotor.22 Se trata de un síndrome genéticamente heterogéneo, donde las mutaciones en el gen FGFR1 se asocian casi siempre a manifestaciones más leves.
Numerosas dismorfias se han asociado a la baja talla, siendo las causas diversas; se ha observado en los siguientes síndromes: Crouzon, Wolf, Síndrome del brazo largo del cromosoma 12, Asskorg, Aspert, Blom, Cockeyne, Conredi o condrodistrofia punctata, Cornelia-Lange, Donohue o Leprechuanismo y en el Síndrome de Ellis-Van Creveld o displasia condro-ectodérmica.
Específicamente, se describe la baja talla postnatal entre las bases clínicas para establecer el diagnóstico del Síndrome de Crouzon, formando parte del cuadro clínico de este síndrome genético.
Por otro lado, se conoce que la deficiencia de GH puede deberse a defectos de los neurotransmisores, a la deficiencia del factor liberador (GHRH), a la deficiencia de la hormona de crecimiento hipofisario, a la deficiencia de producción de somatomedinas, ya sea parcial o completa, así como a la producción de GH biológicamente inactiva, a la resistencia de acción de las somatomedinas u anomalías del receptor de GH.
Se ha señalado que cuando existe hipoplasia hipofisaria asociada a agenesia del cuerpo calloso se puede encontrar un patrón retardado e incluso atenuado de crecimiento desde los dos años de edad; dicho patrón produce estatura final menor a la expresada en la centila III poblacional por decremento en la velocidad de crecimiento a partir de los dos años y menor intensidad y duración del brote de crecimiento puberal.
Resulta interesante que en nuestro paciente la baja talla aparece por una deficiencia de hormona del crecimiento selectiva e idiopática, asociada al síndrome genético: este paciente tuvo dos intervenciones quirúrgicas a las que fue sometido a los 2 y 5 años de edad, pero no requirieron abordaje de la región selar, por lo que no pensamos que pudiera haber sido la causa de la misma. Se ha reportado en la literatura médica pacientes con el Síndrome de Crouzon;25,26 sin embargo, no hemos encontrado otras descripciones similares a las nuestras, ya que en la presentación aquí expuesta está asociado a la deficiencia de hormona de crecimiento, situación no encontrada en la literatura nacional ni en la bibliografía internacional revisada.
A modo de conclusión, consideramos que quizás la heterogeneidad genética alélica y no alélica de locus puede explicar la variabilidad fenotípica intrafamiliar de estas anomalías en el Síndrome de Crouzon.
Por tanto, sugerimos que el seguimiento de estos pacientes sea multidisciplinario, en conjunto con el asesoramiento genético a la familia, donde las decisiones de tratamiento sean individualizadas y determinadas por el tipo y la gravedad de la presentación clínica.
Bibliografía
1. Salas Mamani A. Craneosinostosis simple. Revista de Actualización Clínica. 2014; 46: 2421-2425.
2. Palafox D, Rivas E, Rodríguez D, Queipo G. Malformaciones craneofaciales. Revista Médica del Hospital General de México. 2012;75 (1):50-59.
3. Goyenechea F. Hodelín R. Craneosinostosis. [Sitio de Internet] 2013: [Citado 8 de Diciembre del 2006]. Disponible en: http://neuroc99.sld.cu/text/craneosinostosis.htm
4. Jones KL. Smith’s Recognizable Patterns of Human Malformation. Elsevier Saunders; 2006.
5. Maeda T, Hatakenaka M, Muta H, et al. Clinically mild, atypical, and aged craniofacial syndrome is diagnosed as Crouzon syndrome by identification of a point mutation in the fibroblast growth factor receptor 2 gene (FGFR2) Internal Medicine. 2004;43(5): 432 435. [PubMed].
6. Vidal Sanahujaa A, Gean Molinsb E, Sánchez Garréc C, Quilis Esquerrad J, García Fructuosoe G, Costa Clara JM. Síndrome de Crouzon: a propósito de 2 casos. Entidades craneoestenóticas alélicas de los genes FGFR. An Pediatr (Barc). 2012;77(4):272-278.
7. Craniosynostosis syndromes (FGFR-related). GeneReviews at GeneTests, GeneClinics: Medical Genetics Information Resource [database online]. Seattle: University of Washington; 1997-2002. Disponible en: http://www.genetests.org
8. Sharda S, Panigrahi I, Gupta K, Singhi S, Kumar R. A newborn with acanthosis nigricans: can it be Crouzon syndrome with acanthosis nigricans? Pediatr Dermatol. 2010;27:43-7.
9. Gupta AK, Koley S, Choudhary S, Bhake A, Saoji V, Salodkar A. A rare association of acanthosis nigricans with Crouzon syndrome. Indian J Dermatol Venereol Leprol. 2010;76:65-7.
10. Di Rocco F, Collet C, Legeai-Mallet L, Arnaud E, Le Merrer M, Hadj-Rabia S, et al. Crouzon syndrome with acanthosis nigricans: a case-based update. Child Nerv Syst. 2011;27:349-54.
11. Ahmed I, Afzal A. Diagnosis and evaluation of Crouzon syndrome. J Coll Physicians Surg Pak 2009; 19(5):318-20.
12. Calzada R. Talla Baja. Intersistemas S.A.C.B. 2007, p. 997- 998. México.
13. Carvajal, F, Pérez C. Síndrome de baja talla. Autores Cubanos, Pediatría. Vol. VI, 2011, p. 2510- 2516.
14. Posnick J C, Ruiz RL. The craniofacial disostosis syndromes: Current surgical thinking and future. Cleft Palate Craniofac J 2000; 37:433.
15. Cholley F, Trivin C, Sainte-Rose C, Souberbielle JC, Cinalli G, Brauner R. Disorders of growth and puberty in children with non-tumoral hydrocephalus. Journal of Pediatric Endocrinology and Metabolism. 2001;14(3):319–327. [PubMed].
16. Chiliquinga Villacis S, Agudo Gonzabay B. Síndrome de Apert: caso clínico y revisión. Memoria de Artículos del Primer Congreso de Ciencia y Tecnología UTMACH. 2015: 43-46.
17. Lapunzina P. Aspectos clínicos y genéticos en tallas bajas disarmónicas. Rev Esp Endocrinol Pediatr. 2015; 6 (Suppl): 9-12.
18. Kouga T, Tanoue K, Matsui K. Airway statuses and nasopharyngeal airway use for airway obstruction in syndromic craniosynostosis. Journal of Craniofacial Surgery. 2014;25(3):762-765.
19. Cerrato FE, Nuzzi LC, Theman TA, Taghinia A, Upton J, Labow BI. Upper extremity anomalies in Pfeiffer syndrome and mutational correlations. Plastic and reconstructive surgery. 2014;133(5): 654e-661e.
20. Nur BG, Pehlivanoğlu S, Mıhçı E, Calışkan M, Demir D, Alper OM, et al. Clinicogenetic study of Turkish patients with syndromic craniosynostosis and literature review. Pediatric Neurology. 2014;50(5):482-490.
21. Agochukwu NB, Solomon BD, Muenke M. Hearing loss in syndromic craniosynostoses: introduction and consideration of mechanisms. American Journal of Audiology. 2014;23(2):135-141.
22. Ettinger N, Williams M, Phillips JA. Variable expressivity and clinical heterogeneity can complicate the diagnosis and management of Pfeiffer syndrome. Journal of Craniofacial Surgery. 2013; 24(5): 1829-1832.
23. Martelli Júnior H, Nascimento de Aquino S, Assis Machado R, Lima Leão L, Della Coletta R, Burle-Aguiar MJ. Pfeiffer syndrome: Clinical and genetic findings in five Brazilian families. Med Oral Patol Oral Cir Bucal. 2014;20(1):e52.
24. Kalathia MB, Parikh YN, Dhami MD, Hapani PT. Pfeiffer syndrome. J Pediatr Neurosci. 2014;9(1):85.
25. Colosimo C, Tartaro A, Cama A, TortoriDonati P. The craniosynostoses. In: TortoriDonati P, Rossi A, Biancheri R (eds). Berlin Springer: Pediatric Neuroradiology 2005: p. 1289–1315.
26. Crouzon syndrome. Cleft Palate Foundation Publications [Artículo en línea]. Disponible en: <http://www.cleftline.org/publications/cruozon.htm> [Consulta: 16 de sept. 2005].