Ciencia, Ambiente y Clima, Vol. 2, No. 2, julio-diciembre, 2019 • ISSN: 2636-2317 (impreso) | 2636-2333 (en línea) • Sitio web: https://revistas.intec.edu.do/
APRENDIZAJE DE MÁQUINA Y APRENDIZAJE PROFUNDO EN BIOTECNOLOGÍA: APLICACIONES, IMPACTOS Y DESAFÍOS
Machine learning and deep learning in biotechnology: applications, impacts, and challenges
*Departamento de Biotecnología, Instituto de Ciencias Biológicas, Universidad Federal de Para, Belém, Para, Brasil. Correo-e: edianfranco@ufpa.br
**Departamento de Biotecnología, Instituto de Ciencias Biológicas, Universidad Federal de Para, Belém, Para, Brasil. Correo-e: rommelramos@ufpa.br

Cómo citar: F. Franco, E., & J. Ramos, R. (2019). Aprendizaje de máquina y aprendizaje profundo en biotecnología: aplicaciones, impactos y desafíos. Ciencia, Ambiente y Clima, 2(2), 7-26. Doi: https://doi.org/10.22206/cac.2019.v2i2.pp7-26
Resumen
La bioinformática es un área que ha modificado la forma en que se diseñan y se desarrollan los experimentos e investigaciones de las áreas biológicas. Ha contribuido a la reducción del tiempo y a la economía de recursos, puesto que muchas de las investigaciones son modeladas a través de herramientas de bioinformáticas antes de pasar a las bancadas de los laboratorios. La biotecnología no ha quedado fuera de los alcances de la bioinformática, impactando directamente áreas como el descubrimiento y el desarrollo de fármacos, mejoramiento de cultivos, biorremediación, estudios de la diversidad ambiental, patología molecular, entre otras. Esto se debe, en gran medida, al desarrollo de las tecnologías de secuenciación de alto rendimiento o Next-generation sequencing (NGS), que han generado gran cantidad de datos que deben ser procesados y analizados para producir nuevos conocimientos y descubrimientos. Lo anterior ha promovido que dos áreas de la bioinformática y la ciencia de la computación, machine learning y deep learning, hayan sido utilizadas para el análisis de estos datos. El “aprendizaje de máquina” aplica técnicas que permiten que las computadoras aprendan, mientras que el “aprendizaje profundo” genera modelos de redes neuronales artificiales que intenta imitar el funcionamiento del cerebro humano, permitiéndoles aprender a partir de los datos y mejorar su aprendizaje a través de las experiencias. Estas dos áreas son esenciales para poder identificar, analizar, interpretar y obtener conocimientos de la gran cantidad de datos biológicos (Big biological data). En este trabajo hacemos una revisión de estas dos áreas: el aprendizaje de máquina y el aprendizaje profundo, orientado al impacto y sus aplicaciones en el área de biotecnología.
Palabras clave:
Abstract
Bioinformatics is an area that has changed how experiments and research in biological sciences are designed and developed. This area has contributed to reducing the time and economic resources because many investigations are modeled through bioinformatics tools before moving to the wet laboratory. Biotechnology is an area that has not been developed beyond the scope of bioinformatics, impacted areas such as drug discovery and development, crop improvement, bioremediation, studies of environmental diversity, molecular pathology, and other areas within biotechnology directly. Due to the development of the high throughput sequencing technologies or Nextgeneration sequencing (NGS), a large amount of data is being generated, which needs to be processed and analyzed to generate new knowledge and discoveries. To analyze the amount of data, the fields of machine learning and deep learning from computer science were integrated into bioinformatics. Machine learning applies techniques that allow computers to learn, while deep learning generates models of artificial neural networks that try to mimic the functioning of the human brain allowing them to learn from the data and improve their learning through experiences. These areas are essential to identify, analyze, interpret, and obtain knowledge of a large amount of biological data (Big biological data). In this study, we present a review of these two areas: machine learning and deep learning, impact-oriented, and its applications in the area of biotechnology
Keywords:
1.Introducción
En las últimas décadas, la bioinformática ha sido unos de los ejes fundamentales para el desarrollo de las mayorías de las investigaciones e innovaciones en biología y ambiente. Es un área considerada “nueva” por muchas personas, aunque su historia se remonta a los primeros años de las computadoras (Gauthier, Vincent, Charette, & Derome, 2018). Además, es multidisciplinar, compuesta por la biología, la matemática, la estadística, las ciencias de la computación y la bioestadística, su objetivo es la aplicación de las tecnológicas de la información para manejar, almacenar, sistematizar, analizar e interpretar los datos biológicos (Searls, 2010; SINGH, SINGH, CHAND, & KUSHWAHA, 2011).
La bioinformática ha impactado en gran medida la ciencia biológica, que ha cambiado la forma en la que aborda las investigaciones. Hace algunos años comenzaban en las bancadas de los laboratorios (wet laboratory), lo que suponían una inversión de recursos, en la actualidad, la mayoría de las investigaciones inician en los computadores, lo cuales permiten simular ambiente y realizar experimentos in-silico, para experimentar y validar hipótesis antes de ir a la bancada de los laboratorios, lo que representa una economía de tiempo y recursos (Searls, 2010; SINGH et al., 2011).
Esta área ha cobrado gran importancia en la mayoría de las disciplinas biológicas, debido, en gran medida, al desarrollo y perfeccionamiento de las tecnologías de la secuenciación de alto rendimiento o también llamada de secuenciadores de “Next-generation sequencing” (NGS) (Metzker, 2010).
Las NGS posibilitaron la disminución de los costos y del tiempo necesario para la secuenciación de los genomas, así como el aumento de la confiabilidad de los datos obtenidos (Thermes, 2014). La introducción al mercado de estas tecnologías provocó un incremento en las investigaciones en las áreas omicas, generando un crecimiento exponencial de los datos biológicos y genómicos, además de permitir el estudio de nuevos organismos y especies que antes no habían sido estudiadas. Asimismo, favoreció el surgimiento de nuevos campos de estudios como la metagenómica, epigenética, transcriptómicas, entre otras, que han permitido profundizar en el conocimiento de los organismos el medio ambiente (Behjati & Tarpey, 2013; Mccombie, Mcpherson, & Mardis, 2019; Metzker, 2010; Thermes, 2014).
Debido a esta gran cantidad de datos biológicos (Big biological data) y a la creciente necesidad de analizar y manipular estos datos para obtener nuevos conocimientos la bioinformática se ha constituido en un área esencial dentro de las ciencias biológicas (Gauthier et al., 2018; Min, Lee, & Yoon, 2017).
Dentro de la biotecnología, el impacto de la bioinformática no ha sido menor, convirtiéndose en un apoyo fundamental para la mayoría de los análisis y experimentos que se realizan dentro de esta área. Además, ha contribuido al crecimiento que ha experimentado la biotecnología en los últimos años (Kumar & Chrodia, 2016).
Las herramientas de bioinformática apoyan la biotecnología en áreas como el modelamiento de proteínas, el desarrollo de nuevos medicamentos y vacunas, la mejora de los suelos, eficiencia de la agricultura, biorremediación (figura 1), además de ayudar en la
comprensión de los genes y de la complejidad de los genomas de muchos organismos que son de interés para las áreas biotecnológicas mediante el uso de las bases de datos biológicas y las herramientas bioinformáticas (Bansal, 2005; de Carvalho et al., 2019; Kumar & Chrodia, 2016).
Con la finalidad de poder procesar y obtener mayores conocimientos de la gran cantidad de datos biológicos disponibles, nuevas técnicas y metodologías de bioinformáticas y las ciencias de la computación han sido aplicada al área de biotecnología, ejemplo de ello: el aprendizaje de máquina y redes neurales de aprendizaje profundo, las cuales permiten el procesamiento de grandes conjuntos de datos biológicos y la obtención de nuevos descubrimientos y conocimientos, que con otras técnicas no habrían sido posibles.
Este trabajo tiene como objetivo hacer una revisión del impacto del aprendizaje de máquina y el aprendizaje profundo en el área de biotecnología, así como las herramientas utilizadas, a fin de ofrecer una perspectiva de estas metodologías y sus aplicaciones.

1-Aprendizaje de máquina (Machine Learning)
El aprendizaje de máquina (AM) es un área de la ciencia de la computación que se dedica al desarrollo y aplicación de técnicas y algoritmos computacionales, capaces de “aprender” y de perfeccionar sus aprendizajes a través de las experiencias y adaptación a las condiciones que cambian con el tiempo, de esta forma aumentan el rendimiento de las máquinas y su capacidad de inferir y obtener nuevos conocimientos de grandes conjuntos de datos. (Dixit & Prajapati, 2015; Libbrecht & Noble, 2015).
Uno de los objetivos principales de esta área es que los computadores aprendan; para este fin se utilizan diferente técnicas y metodologías estadísticas, matemáticas y lógicas, que posibilitan que las técnicas de AM puedan manejar e interpretar grandes cantidades de datos. AM se ha convertido en un área atractiva para la biotecnología, debido a su adaptación a los diversos tipos de análisis y datos, así como la diversidad de experimentos que se pueden realizar utilizando estos algoritmos.
Estas técnicas se pueden clasificar en tres tipos: supervisados, no supervisados y semi-supervisados, siendo que cada uno tiene una función diferente según el tipo de datos con el que se trabaja y el objetivo de la investigación o análisis que se está realizando.
1.1 Aprendizaje de máquinas supervisadas
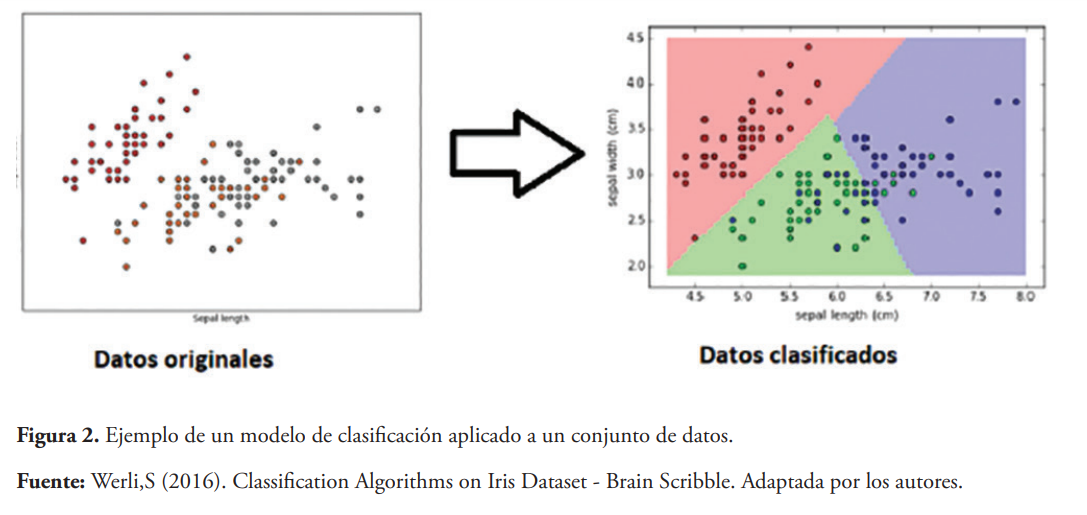
Las técnicas de aprendizaje supervisado o técnicas predictivas están basadas en algoritmos que necesitan conocimientos previos (datos etiquetados) para ser entrenados (conjunto de entrenamiento). A partir de este entrenamiento son capaces de predecir la etiqueta de un nuevo dato de entrada sin etiqueta (conjunto de prueba) (Figura 2) (Libbrecht & Noble, 2015). Un ejemplo de este tipo de clasificación es la predicción biomarcadores de tejidos específicos a partir de la expresión génica (Mamoshina, Vieira, Putin, & Zhavoronkov, 2016a).
Dentro de estas técnicas se encuentran diferentes tipos de algoritmos como son: las máquinas de vectores de soporte (SVM), las cuales construyen líneas de separaciones para distinguir entre dos objetos diferentes dentro de un espacio multidimensional (Noble, 2006); los árboles de decisiones, que crea reglas de división dentro de los conjuntos de datos y las organiza en forma de árbol, para de este modo poder predecir el valor de una variable partiendo de otras variables de entradas (Lavecchia, 2015); otro tipo de algoritmos son los basados en instancias o de aprendizaje vago, que clasifican un nuevo valor de entrada con base en la proximidad de este valor con los valores que fueron utilizados para el entrenamiento de los modelos (Lavecchia, 2015).
Otra técnica del aprendizaje de máquina, que es clasificada como supervisada, es algoritmos de regresión, este tipo de algoritmo, a diferencia de los de clasificación, utiliza los datos etiquetados para predecir valores reales como la expresión de un gen, el tamaño de genoma, por ejemplo (Libbrecht & Noble, 2015; Witten, Frank, & Hall, 2011a).
Dentro de las regresiones podemos encontrar los algoritmos de regresión lineal, que permiten identificar la relación de dependencia entre una variable dependiente y otra independiente (Witten, Frank, & Hall, 2011b).
1.2 Aprendizaje de máquinas no supervisadas
Este tipo de aprendizaje está basado en algoritmos que no requieren un conocimiento previo de los datos o datos etiquetados. Este tipo de aprendizaje también es conocido como aprendizaje descriptivo, ya que permite obtener patrones y conocimientos a partir de las características intrínsecas de los datos. Estos algoritmos pueden ser utilizados para identificar grupos de genes con patrones de expresión similares dentro de los datos de expresión génica en diferentes tipos de estreses ambientales (Libbrecht & Noble, 2017).
Dentro de este tipo de aprendizaje podemos encontrar varias metodologías como son las técnicas de agrupamiento (clustering), análisis de componentes principales, selección de variables y reglas de asociaciones.
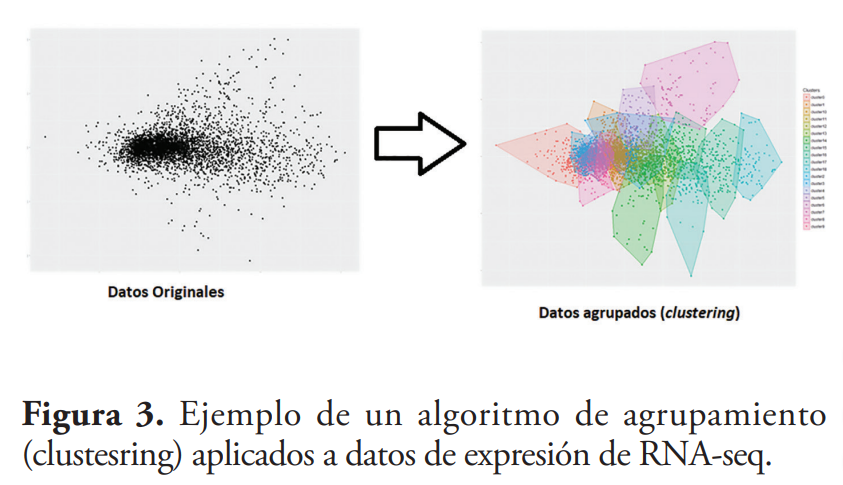
Las técnicas de agrupamiento buscan identificar la estructura de los grupos a través de la información inherente de los datos, atendiendo a la mayor similitud dentro de las miembros que forman los grupos (intra-clusters) y la mayor diferencia entre los miembros que pertenecen a otros grupos (Figura 3). Las técnicas de agrupamientos permiten la identificación de grupos con funciones genéticas similares, así como la identificación de grupos de genes o proteínas con homología, que son importantes para el diseño de vacunas y desarrollo de fármacos. Este tipo de algoritmos han ayudado a la mejora de los cultivos y de la agricultura (Datta & Datta, 2006; Liakos, Busato, Moshou, Pearson, & Bochtis, 2018; Oyelade et al., 2016).
El análisis de componentes principales (PCA) es una técnica utilizada para describir y reducir la dimensionalidad de los conjuntos datos, manteniendo la mayor variabilidad dentro de los datos. Esta técnica tiene la capacidad de identificar tendencia dentro de los conjuntos de datos, lo cual permite identificar y evaluar patrones, similitudes y agrupamiento de forma visual. PCA es mayormente utilizada para reducir la cantidad de datos necesarios para los análisis, manteniendo la representatividad dentro del conjunto y de esta forma poder mejorar el desempeño computacional (Peres-Neto, Jackson, & Somers, 2005; Ringnér, 2008).
La selección de variables permite escoger los atributos o variables que son más significativos para ser utilizados en las construcciones de los modelos y análisis. Esta técnica permite reducir la cantidad de datos presentes en un conjunto, con la finalidad de simplificar la interpretación de los modelos, mejorar el desempeño de los computadores, además de reducir el tiempo de los análisis (James, Witten, Hastie, & Tibshirani, 2013). Estas técnicas son utilizadas cuando tenemos un conjunto de datos de una significativa cantidad de variables, como es el caso de los metagenomas, donde las dimensiones de los datos son significativamente grandes (Al-Ajlan & El Allali, 2018; Bermingham et al., 2015).
Las reglas de asociación son un método que permite identificar relaciones existentes entre las variables de un conjunto de dados, a través de la definición de reglas asociativas. Este tipo de análisis se ha utilizados para la detección y descripción de patrones de relación ocultos dentro de los datos de expresión génica, además para estudiar la regulación génica entre los genotipos y fenotipos (Chen, Tsai, Chung, & Li, 2015; Martínez, Pasquier, & Pasquier, 2008).
1.3 Aprendizaje de máquina semi-supervisado y ensamblado
Las técnicas pertenecientes a este tipo de aprendizaje de máquina son un híbrido entre las dos técnicas anteriores, estos algoritmos reciben un conjunto de datos, una parte de los etiquetados y otras sin etiquetar, para construir modelos de descripción y predicción de los datos. Estas metodologías son utilizadas para entrenar los sistemas de predicción de genes y mejorar los procesos de anotación automática de los nuevos genomas (Libbrecht & Noble, 2017).
Las técnicas de aprendizaje ensamblados, combinan diferentes algoritmos y técnicas independientes en un solo modelo, con la finalidad de mejorar la predicción de los algoritmos. Este tipo de metodologías se basa en el hecho de que las diferentes técnicas presentan algún tipo de debilidad o sesgos para identificar patrones específicos o trabajar con un tipo datos. Mediante la combinación diferentes tipos de algoritmos se pueden obtener modelos predictivos más sólidos, en comparación con utilización de un solo un algoritmo (Camacho, Collins, Powers, Costello, & Collins, 2018; Libbrecht & Noble, 2017).
1.4 Herramientas utilizadas para el aprendizaje de máquina
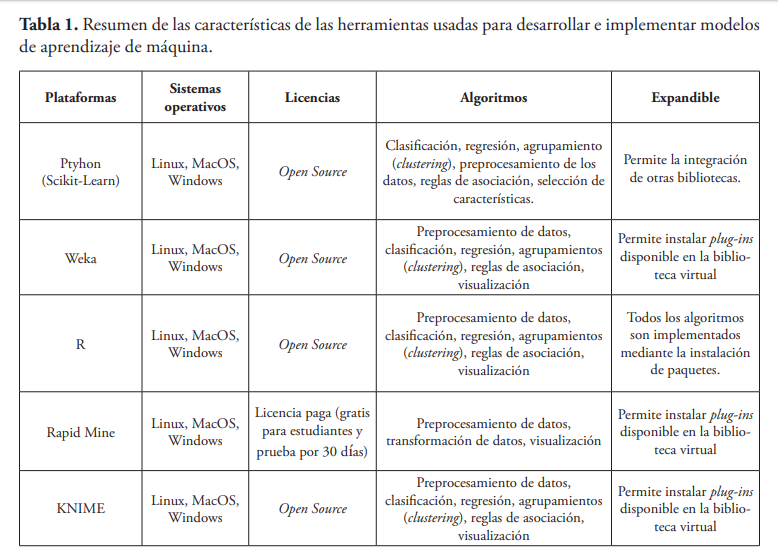
Para desarrollar y ejecutar modelos de aprendizaje de máquina existen diferentes herramientas y lenguajes de programación: Python (Scikit-Learn), GNU R, Weka y Rapidminder. La mayoría de estas herramientas son open-source (software libre), ofrecen su utilización de forma gratuita por los usuarios, además de ser multiplataformas, permitiendo que puedan ser ejecutadas en la mayoría de los sistemas operativos presentes en el mercado. La Tabla 1 muestra un resumen de las principales características de estas herramientas.
Python es uno de los leguajes de programación más utilizados dentro de las áreas científicas y la ciencia de datos, debido a su versatilidad, la sintaxis simple, además es multiplataformas, lo que permite que los programas desarrollados en este lenguaje se puedan ejecutar en Linux, MacOS y Windows. Este lenguaje de programación permite el uso de bibliotecas, que son un conjunto de funciones (programas) con una finalidad específica y predeterminada.
Una de las bibliotecas más utilizadas dentro del área aprendizaje de máquina en Python es la Scikit-Learn, esta biblioteca permite la implementación de algoritmos supervisados y no supervisados además de la visualización de los datos al combinarse con otras bibliotecas (Pedregosa et al., 2011).
Weka es un software desarrollado en Java, que permite la construcción e implementación de modelos y algoritmos de aprendizaje de máquina, sin la necesidad de tener muchos conocimientos previos en el área o de manejar un lenguaje de programación (Hall et al., 2009). Esta herramienta es muy utilizada dentro de las áreas de genómica y bioinformática para realizar análisis de grandes conjuntos de datos (Frank, Hall, Trigg, Holmes, & Witten, 2004). Las últimas versiones de Weka permiten la instalación de plug-in adaptado a la necesidad de los usuarios y que están disponible en un repositorio online. Esta herramienta también permite la integración con leguaje de programación como son Python y R (Beckham, Hall, & Frank, 2016).
GNU R o simplemente R es un lenguaje y entorno de programación con orientación estadística, que permite realizar diversos tipos análisis y experimento debido a la cantidad de paquetes, con funciones y características predefinidas, disponibles que se adaptan a las diversas necesidades y áreas de conocimiento de los usuarios (Free Software Foundation, 2016). Las técnicas de aprendizaje de máquina en R se puede implementar a través de diferentes paquetes entre los que podemos encontrar: CARET (clasificación y regresión) (Kuhn, 2008), randomForest (árboles de decisiones) (Liaw & Wiener, 2002), Kernlab (SVM) (Karatzoglou, Smola, Hornik, & Zeileis, 2004), BayesTRee (árboles de decisiones basados en modelos bayesianos) (Chipman, George, & McCulloch, 2010), y 1071 (clustering) (Meyer, Dimitriadou, Hornik, Weingessel, & Leisch, 2017), Rweka (integración con Weka) (Hornik, Buchta, & Zeileis, 2009).
RapidMiner es una plataforma para el análisis de grandes conjuntos de datos a través de técnicas de aprendizaje de máquina y análisis de datos. Este programa brinda una interfaz gráfica de fácil uso y que no requiere conocimientos previos de programación. Permite encadenar diferentes métodos y procesos para el análisis y visualización de los datos. Las funcionalidades de esta herramienta pueden ser expandidas mediante la instalación de plug-ins para realizar análisis específicos (Mierswa, Wurst, Klinkenberg, Scholz, & Euler, 2006).
Otra herramienta utilizada para el área de aprendizaje de máquina es KMINE, fue inicialmente desarrollada para análisis bioinformáticos, en la actualidad es usada por diversas áreas de la ciencia y los negocios (Tiwari & Sekhar, 2007). Ofrece una interfaz gráfica de fácil uso, la cual permite el análisis de los datos a través de diagramas de procesos donde se pueden agregar diferentes tipos de algoritmos y modelos que permiten el pre-procesamiento, procesamiento y visualización de los datos (Berthold et al., 2006).
1.5 Aprendizaje de máquina aplicado a la biotecnología
Las diversas técnicas que brinda el área de aprendizaje de máquinas han sido aplicadas en diferente estudios e investigaciones dentro de las áreas de la biotecnología, como son biorremediación, descubrimiento y desarrollo de agricultura, mejoramiento de cultivos, estudio de diversidad ambiental, entre otros.
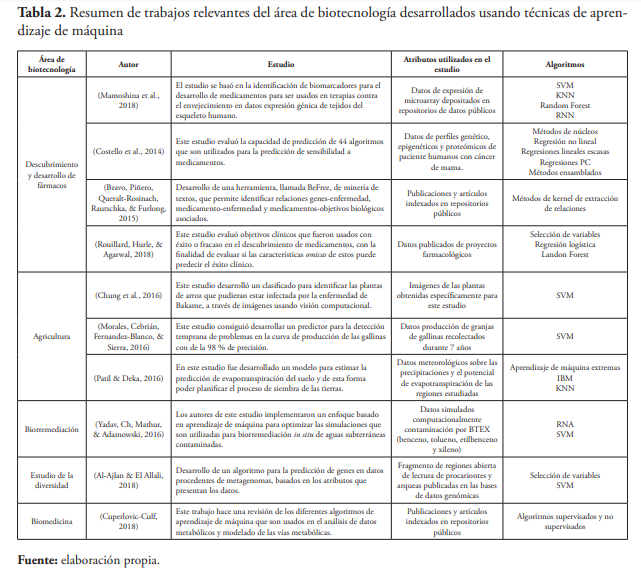
La tabla 2 muestra un resumen de algunos estudios que han sido desarrollados usando técnicas de aprendizaje de máquina en diversas áreas de la biotecnología.
2. Aprendizaje profundo (Deep Learning)
Una de las áreas “aprendizaje máquina” que más ha llamado la atención de la comunidad científica en los últimos años, debido a aplicabilidad en diferentes áreas y contextos de la ciencia es el aprendizaje profundo (AP). Esta es una clase de AM que permite la extracción de abstracciones de alto nivel de grandes conjuntos de datos brutos, con alta dimensionalidad y heterogeneidad (Eraslan, Avsec, Gagneur, & Theis, 2019; Mamoshina, Vieira, Putin, & Zhavoronkov, 2016b).
Los modelos desarrollados técnicas de AP, permiten predecir procesos celulares, variaciones genéticas, procesos de co-expresión celular y regulación génica, además que ayudan en el descubrimiento y desarrollo de nuevos fármacos y el estudio de la biodiversidad, entre otras áreas de la biotecnología (Eraslan et al., 2019; Wainberg, Merico, Delong, & Frey, 2018).
AP ha impactado las investigaciones en las áreas biológicas, genética, genómica, biotecnología y biomédica debido a la capacidad de poder manejar e integrar grandes cantidades de datos, como los que son generado en estas áreas, y a partir de estos poder aprender e identificar relaciones complejas dentro de los datos que permiten producir nuevos conocimientos (Wainberg et al., 2018).
El AP se basa en la aplicación de algoritmos de redes neuronales artificiales (RNA) de varias capas que son capaces de encontrar e inferir patrones complejos dentro de los datos. Los algoritmos de RNA intentan reproducir el comportamiento del cerebro humano, mediante la reproducción de los procesos de conexión de las neuronas, por esta razón estas redes han sido hábiles a la hora de aprender tareas y funciones complejas a partir de las entradas que reciben, además de que consiguen adaptarse a los cambios que pueden sufrir las entradas (Y. Park & Kellis, 2015; Wainberg et al., 2018).
Las RNA son redes completamente conectadas, donde una neurona que está en una capa específica está complemente conectada con la neurona de la siguiente capa a través de un enlace (Figura 4). El comportamiento de la RNA es definido por la fuerza, las conexiones entre la neurona y la forma en que las entradas se transforman mediante función de activación predefinida (sigmoide o tangente hiperbólica) que determina un valor o peso; y este valor, la activación o desactivación de la siguiente neurona dentro de la red. Según la cantidad de capas y neuronas la red puede tener diferentes capacidades de profundizar en aprendizaje y de autorregulación (Van Gerven & Bohte, 2017).
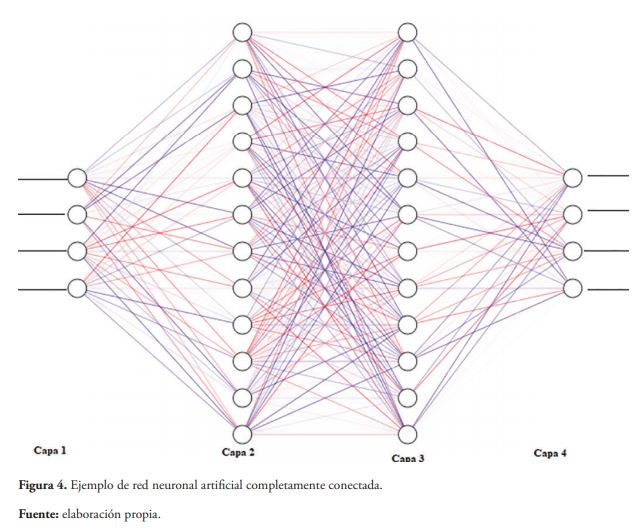
El desarrollo de las RNA y el aprendizaje profundo ha sido posible debido al aumento de la capacidad computacional que se ha experimentos en las últimas décadas, así como desarrollo de las unidades de procesamiento gráficos (GPU) que han aumentado la capacidad de procesamiento de los equipos y permitido el desarrollo de redes neuronales artificiales más robustas (Eraslan et al., 2019).
Los algoritmos o técnicas de aprendizaje profundo, al igual en aprendizaje de máquina, se puede clasificar en supervisados y no supervisados, según su capacidad de predecir o describir los grandes conjuntos de datos.
2.1 Aprendizaje profundo supervisado
El objetivo principal de este tipo de aprendizaje es construir redes (modelos) que reciban un conjunto de variables de entrada y devuelva una predicción de una variable objetivo (Eraslan et al., 2019). Un ejemplo de este tipo de aprendizaje es la predicción de marcos abiertos de lecturas en genomas.
Varios tipos de algoritmos de redes neuronales son clasificadas como aprendizaje supervisados: red neuronal convolucional, red neuronal recuente y rede neuronal convolucional gráfica.
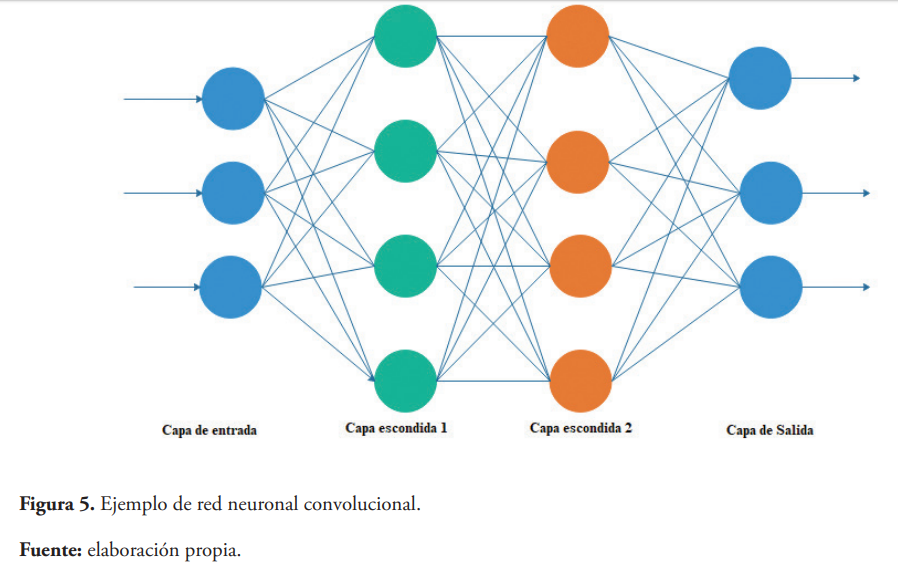
Las redes neuronales convolucionales son redes neuronales completamente conectadas que utilizan matrices bidimensionales, llamadas ventanas, para realizar mapeo de los datos, intentado imitar las neuronas de las cortezas visual de cerebro humano (Figura 5) (Eraslan et al., 2019; Wainberg et al., 2018). Estas redes han sido utilizadas para la clasificación de los sitios de unión de factores de transcripción (Zou et al., 2016); Wang, Tai, E, & Wei, 2018), la predicción de fenotipos moleculares (Kelley et al., 2018), metilación de ADN (Zhou et al., 2018), análisis de la expresión génica y microARN (Budach & Marsico, 2018).
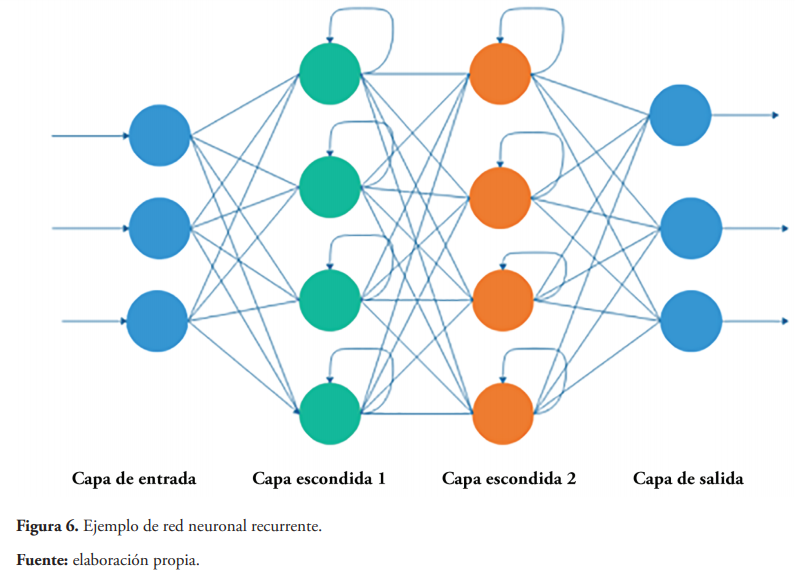
Las redes neuronales recurrentes son utilizadas cuando se trabaja con datos dinámicos que pueden cambiar en el tiempo. Este tipo de redes reciben un retorno que permite la retroalimentación de la red, lo que mejora el rendimiento en la detección de patrones de los datos (Figura 6) (Eraslan et al., 2019). Dentro de las áreas biológicas estas redes han sido aplicadas para predecir el estado de metilación
del ADN de las células (Angermueller, Lee, Reik, & Stegle, 2017) y para la predicción de sitio de factores de transcripción, además que han permitido mejorar la accesibilidad a la información del ADN (Pan, Rijnbeek, Yan, & Shen, 2018), otra área en la que este tipo de redes han sido utilizadas, en la predicción de modelos de interacciones de microARN-miroARN (S. Park, Min, Choi, & Yoon, 2016).
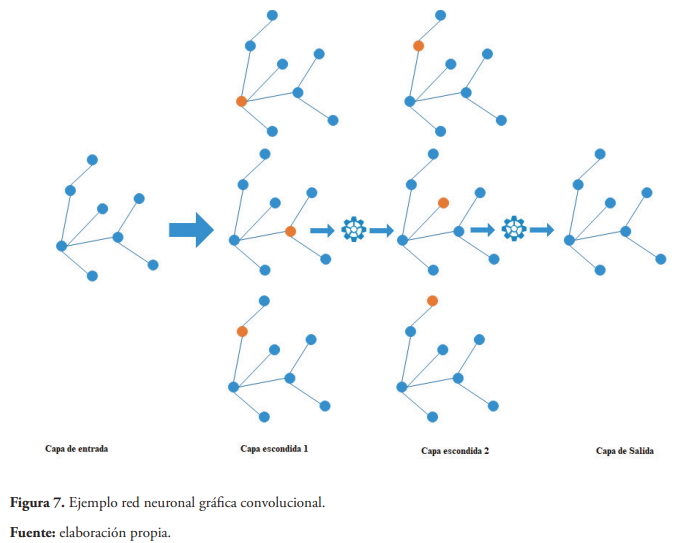
Las redes neuronales gráficas convolucionales, utilizan gráficos para aprender los patrones de los datos, utilizando los nudos y las conexiones de estos nudos para resolver los problemas de aprendizaje automático. Para aprender estas redes aplican múltiples transformaciones a las capas que forma el gráfico, en cada una de estas trasformaciones agregan características a los nudos y sus vértices, permitiendo que la red pueda obtener nuevos conocimientos sobre los datos (Figura 7) (Eraslan et al., 2019). En las áreas de genómica y bioinformática este tipo de redes han sido utilizadas para la predicción de la expresión génica dada la expresión de otros genes (Dutil, Cohen, Weiss, Derevyanko, & Bengio, 2018), como en la clasificación de diferentes subtipos de cáncer (Rhee, Seo, & Kim, 2017).
2.2 Aprendizaje profundo no supervisando
El objetivo principal de este tipo de aprendizaje es caracterizar y describir los conjuntos de datos no etiquetados, permitiendo descubrir y aprender sus patrones y características, de esta forma poder entender las relaciones existen dentro de estos datos (Eraslan et al., 2019).
Dentro del aprendizaje profundo podemos encontrar algoritmos o modelos como autoencode y las redes generativas antagónicas, estos son utilizados para describir los grandes conjuntos de datos.
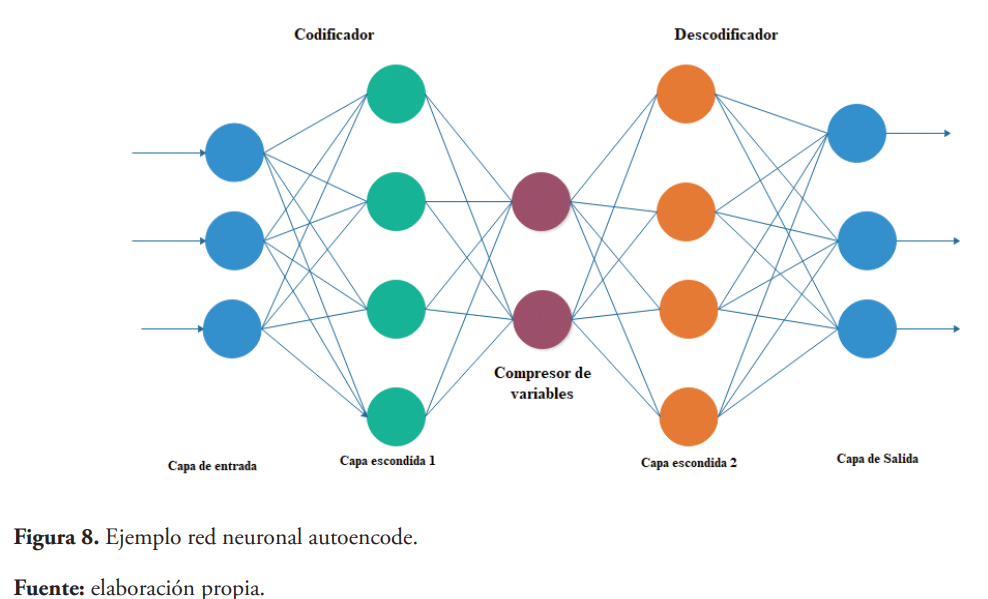
Autoencode es un algoritmo de red neuronal, que trata de aprender parámetros y características que le permiten producir una salida lo más próxima a la entrada recibida, pero con dimensiones reducidas. Este algoritmo utiliza una capa oculta de denominada de cuello de botella que le permite reducir las dimensiones de los datos recibidos sin pérdidas significativas de representatividad de los mismos (Figura 8) (Angermueller, Pärnamaa, Parts, & Stegle, 2016; Eraslan et al., 2019; LeCun et al., 2015). Redes basadas en este algoritmo han sido utilizadas para imputar datos perdidos en conjuntos de datos (Scholz, Kaplan, Guy, Kopka, & Selbig, 2005), extracción de patrones de expresión génica (Tan et al., 2017; Tan, Hammond, Hogan, & Greene, 2016) y detención de valores atípicos dentro de los datos genómicos y transcriptómicos (Brechtmann et al., 2018).
Unos de los algoritmos más recientes dentro del aprendizaje profundo son las redes generativas antagónica que fueron introducidas en 2014 por un ingeniero de Google (Goodfellow et al., 2014). Estas redes consiguen producir salidas realistas, que son generadas a partir de los datos de entradas; por ejemplo, producen una imagen completamente nueva a partir de la inserción de varias imágenes dentro de la red, además logra discriminar si un elemento de la red es real o fue generado por la red. Para regenerar estos resultados este algoritmo utiliza un modelado degenerativo que está compuesto por dos redes neuronales, una red generadora y un discriminador, que se entrenan conjuntamente. El objetivo de la red generadora es generar datos que son realista e intenta engañar a la discriminadora; por otro lado, la red discriminadora identifica y clasifica si el elemento dado es real o fue generado por la red generadora (Eraslan et al., 2019; Goodfellow et al., 2014). Esta red se ha utilizado para originar secuencia de ADN que codifican aminoácidos, para el diseño de sonda de ADN que en experimentos de expresión de microarray (Gupta & Zou, 2018). Por ser relativamente nueva todavía no se ha explotado toda el potencial y la capacidad de aprendizaje de este tipo de red.
1.5 Aprendizaje de máquina aplicado a la biotecnología
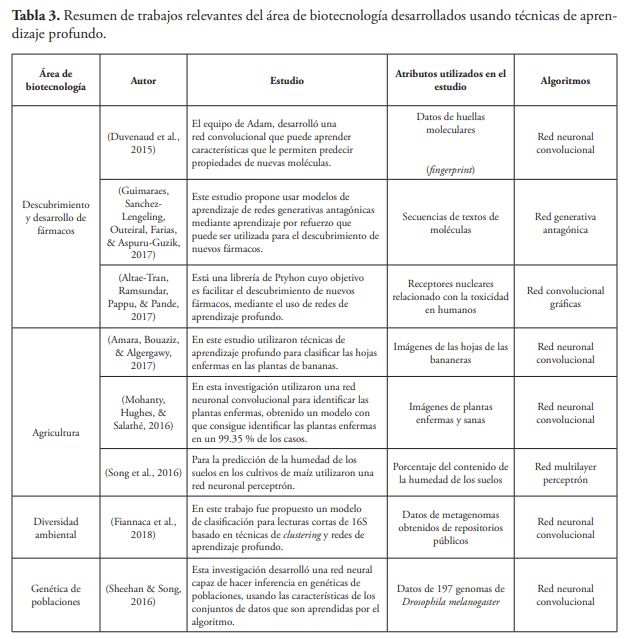
Las redes neuronales artificiales han sido aplicadas para solucionar diferentes problemas dentro del área de biotecnología, desde el descubrimiento y desarrollo de nuevos fármacos hasta el análisis de suelo, para predecir la humedad y el mejor periodo para la siembra. La tabla 3 muestra un resumen de trabajos biotecnológicos relevantes, desarrollados aplicando la técnica de aprendizaje profundo.
2.3 Herramientas utilizadas para aprendizaje de aprendizaje profundo
Varias herramientas o frameworks han permitido el desarrollo y la implementación de códigos y programas de aprendizaje profundo, entre los que pueden ser citados: TensorFlow (Abadi, Barham, et al., 2016), Keras (Chollet, 2015) y PyTorch (Paszke et al., 2017).
TensorFlow es una biblioteca para desarrollar, probar y ejecutar los códigos y programas de aprendizaje profundo desarrollado por Google. En la actualidad, es una de las interfaces más utilizadas por los programadores, debido a que esta se puede ejecutar en diversos sistemas operativos y dispositivos, además que puede trabajar con unidades centrales de procesamiento (CPU) y GPUs sin que el programador necesite hacer grandes cambios en los códigos. Con esta herramienta se pueden implementar algoritmos de aprendizaje profundo supervisado y no supervisados. TensorFlow es una herramienta open-source que ha sido muy utilizada en el desarrollo de varias soluciones dentro de las áreas biológicas y de biotecnología (Abadi, Agarwal, et al., 2016).
Otra de las herramientas de gran importancia para el desarrollo de soluciones de aprendizaje profundo es Keras, una interfaz de programación de aplicaciones (API), que permite el desarrollo, prueba y ejecución del diferentes algoritmos y bibliotecas de aprendizaje profundo como son TensorFlow, CNTK (Seide & Agarwal, 2016) o Theano (Bergstra et al., 2010). Esta API fue desarrollada con el objetivo de permitir una implementación fácil y rápida de los algoritmos, modelos y prototipos de aprendizaje profundo supervisados y no supervisados, además que puede trabajar de forma eficiente en equipos que funcionen con CPU o GPU (Chollet, 2015).
PyTorch es una biblioteca de Python que permite desarrollar aplicaciones y códigos de aprendizaje profundo, esta fue desarrollada por Facebook, siendo en la actualidad una de las principales herramientas para el desarrollo a computación visual y procesamiento del leguaje natural. Esta biblioteca es open-source, lo que permite que pueda ser usada por diferentes usuarios de diversas áreas científicas (Paszke et al., 2017).
Conclusión
La aplicación de técnicas de bioinformática y ciencia de la computación en las áreas biológicas han revolucionado la forma de hacer ciencia, estas han permitido que grupos de investigaciones, grandes y pequeños, puedan tener acceso a las mismas oportunidades, que ya no necesitan sofisticados laboratorios para realizar los análisis o experimentos, debido a la facilidad con que los datos son obtenidos, generados, procesados y almacenados, mediante diferentes técnicas que pueden ser desarrolladas y ejecutadas en pequeñas computadoras de escritorio o grandes servidores. La entrada al mercado de las tecnologías de nueva secuenciación de alto rendimientos o NGS provocó la disminución de los costos de secuenciación, lo que dio como resultado un aumento significativo de la cantidad de datos genómicos que está siendo generada cada día. La adopción de las técnicas “aprendizaje de máquina” y “aprendizaje profundo” ha permitido interpretar y obtener nuevos conocimientos de forma eficiente y rápida de estos grandes conjuntos de datos.
Las técnicas de AM y AP han permito el desarrollo de aplicaciones y programas bioinformáticos que están siendo usados por los biotecnólogos y biólogos para realizar diversos análisis en sus respectivas áreas de conocimientos. Estas técnicas han impactado de forma significativa el descubrimiento y desarrollo de fármacos, medicina personalizada, oncología, estudio de la diversidad ambiental, mejora de cultivos, entre otros, que tienen gran impacto económico. Además, han incentivado el surgimiento de compañías de matriz biotecnológica, que basan sus procesos e investigaciones en este tipo de técnicas.
En el futuro estas técnicas van a continuar tomando mayor relevancia dentro de las áreas de la biotecnología, debido a que existen investigaciones que están trabajando en proceso de integración de los datos ómicos, lo que van a permitir que se pueden usar datos de diferentes tipos y orígenes, permitiendo que se puedan hacer nuevos descubrimientos sobre las relaciones e interacciones entre los diferentes procesos biológicos que suceden dentro de los organismos, mejorando los conocimientos sobre los procesos que sustentan la vida.
Las herramientas y técnicas bioinformáticas van a continuar ganando espacio dentro de las ciencias biológicas, debido a la magnitud y complejidad de los datos que estas áreas generan, lo que asegura que el AM y el AP se conviertan en herramientas cotidianas para los análisis y experimentos.
Agradecimientos
Este trabajo fue desarrollado gracias al apoyo de la Coordenação de aperfeiçoamento de pessoal de nivel superior-CAPES, Brasil y al Consejo Nacional de Desarrollo Científico y Tecnológico-CNPQ Brasil.
Referencias
Abadi, M., Agarwal, A., Barham, P., Brevdo, E., Chen, Z., Citro, C., … Devin, M. (2016). Tensorflow: Large-scale machine learning on heterogeneous distributed systems. ArXiv Preprint ArXiv: 1603.04467.
Abadi, M., Barham, P., Chen, J., Chen, Z., Davis, A., Dean, J., … Zheng, X. (2016). TensorFlow : A System for Large-Scale Machine Learning This paper is included in the Proceedings of the TensorFlow : A system for large-scale machine learning.
Al-Ajlan, A., & El Allali, A. (2018). Feature selection for gene prediction in metagenomic fragments. BioData Mining, 11(1), 9. https://doi.org/10.1186/s13040-018-0170-z
Altae-Tran, H., Ramsundar, B., Pappu, A. S., & Pande, V. (2017). Low data drug discovery with one-shot learning. ACS Central Science, 3(4), 283–293.
Amara, J., Bouaziz, B., & Algergawy, A. (2017). A Deep Learning-based Approach for Banana Leaf Diseases Classification. In BTW (Workshops) (pp. 79–88).
Angermueller, C., Lee, H. J., Reik, W., & Stegle, O. (2017). DeepCpG: accurate prediction of single-cell DNA methylation states using deep learning. Genome Biology, 18(1), 67.
Angermueller, C., Pärnamaa, T., Parts, L., & Stegle, O. (2016). Deep learning for computational biology. Molecular Systems Biology, 12(7), 878. https://doi.org/10.15252/msb.20156651
Bansal, A. K. (2005). Bioinformatics in microbial biotechnology - A mini review. Microbial Cell Factories, 4(ii), 1–11. https://doi.org/10.1186/1475-2859-4-19
Beckham, C., Hall, M., & Frank, E. (2016). WekaPyScript: Classification, Regression, and Filter Schemes for WEKA Implemented in Python. Journal of Open Research Software, 4. https://doi.org/10.5334/jors.108
Behjati, S., & Tarpey, P. S. (2013). What is next generation sequencing? Archives of Disease in Childhood: Education and Practice Edition, 98(6), 236–238. https://doi.org/10.1136/archdischild-2013-304340
Bergstra, J., Breuleux, O., Bastien, F., Lamblin, P., Pascanu, R., Desjardins, G., … Bengio, Y. (2010). Theano: a CPU and GPU math expression compiler. In Proceedings of the Python for scientific computing conference (SciPy) (Vol. 4). Austin, TX.
Bermingham, M. L., Pong-Wong, R., Spiliopoulou, A., Hayward, C., Rudan, I., Campbell, H., … Haley, C. S. (2015). Application of high-dimensional feature selection: evaluation for genomic prediction in man. Scientific Reports, 5, 10312. https://doi.org/10.1038/srep10312
Berthold, M. R., Cebron, N., Dill, F., Di Fatta, G., Gabriel, T. R., Georg, F., … Wiswedel, B. (2006). KNIME: The konstanz information miner. 4th International Industrial Simulation Conference 2006, ISC 2006, 11(1), 58–61.
Bravo, À., Piñero, J., Queralt-Rosinach, N., Rautschka, M., & Furlong, L. I. (2015). Extraction of relations between genes and diseases from text and large-scale data analysis: Implications for translational research. BMC Bioinformatics, 16(1), 1–17. https://doi.org/10.1186/s12859-015-0472-9
Brechtmann, F., Mertes, C., Matusevičiūtė, A., Yepez, V. A., Avsec, Ž., Herzog, M., … Gagneur, J. (2018). OUTRIDER: A statistical method for detecting aberrantly expressed genes in RNA sequencing data. The American Journal of Human Genetics, 103(6), 907–917.
Budach, S., & Marsico, A. (2018). pysster: classification of biological sequences by learning sequence and structure motifs with convolutional neural networks. Bioinformatics, 34(17), 3035–3037.
Camacho, D. M., Collins, K. M., Powers, R. K., Costello, J. C., & Collins, J. J. (2018). NextGeneration Machine Learning for Biological Networks. Cell, 173(7), 1581–1592. https://doi.org/10.1016/j.cell.2018.05.015
Chen, S.-C., Tsai, T.-H., Chung, C.-H., & Li, W.-H. (2015). Dynamic association rules for gene expression data analysis. BMC Genomics, 16(1), 786. https://doi.org/10.1186/s12864-015-1970-x
Chipman, H. A., George, E. I., & McCulloch, R. E. (2010). BART: Bayesian additive regression trees. The Annals of Applied Statistics, 4(1), 266–298.
Chollet, F. (2015). Keras. GitHub. Retrieved from https://github.com/fchollet/keras
Chung, C. L., Huang, K. J., Chen, S. Y., Lai, M. H., Chen, Y. C., & Kuo, Y. F. (2016). Detecting Bakanae disease in rice seedlings by machine vision. Computers and Electronics in Agriculture. https://doi.org/10.1016/j.compag.2016.01.008
Costello, J. C., Heiser, L. M., Georgii, E., Gönen, M., Menden, M. P., Wang, N. J., … Van Westen, G. J. P. (2014). A community effort to assess and improve drug sensitivity prediction algorithms. Nature Biotechnology, 32(12), 1202–1212. https://doi.org/10.1038/nbt.2877
Cuperlovic-Culf, M. (2018). Machine learning methods for analysis of metabolic data and metabolic pathway modeling. Metabolites, 8(1). https://doi.org/10.3390/metabo8010004
Datta, S. S., & Datta, S. S. (2006). Methods for evaluating clustering algorithms for gene expression data using a reference set of functional classes. BMC Bioinformatics, 7, 397. https://doi.org/10.1186/1471-2105-7-397
de Carvalho, L. M., Borelli, G., Camargo, A. P., de Assis, M. A., de Ferraz, S. M. F., Fiamenghi, M. B., … Carazzolle, M. F. (2019). Bioinformatics applied to biotechnology: A review towards bioenergy research. Biomass and Bioenergy, 123(March 2018), 195–224. https://doi.org/10.1016/j.biombioe.2019.02.016
Dixit, P., & Prajapati, G. I. (2015). Machine learning in bioinformatics: A novel approach for DNA sequencing. International Conference on Advanced Computing and Communication Technologies, ACCT, 2015-April, 41–47.
Dutil, F., Cohen, J. P., Weiss, M., Derevyanko, G., & Bengio, Y. (2018). Towards gene expression convolutions using gene interaction graphs. ArXiv Preprint ArXiv:1806.06975
Duvenaud, D. K., Maclaurin, D., Iparraguirre, J., Bombarell, R., Hirzel, T., Aspuru-Guzik, A., & Adams, R. P. (2015). Convolutional networks on graphs for learning molecular fingerprints. In Advances in neural information processing systems (pp. 2224–2232).
Eraslan, G., Avsec, Ž., Gagneur, J., & Theis, F. J. (2019). Deep learning: new computational modelling techniques for genomics. Nature Reviews Genetics, 20(7), 389–403. https://doi.org/10.1038/s41576-019-0122-6
Fiannaca, A., La Paglia, L., La Rosa, M., Renda, G., Rizzo, R., Gaglio, S., & Urso, A. (2018). Deep learning models for bacteria taxonomic classification of metagenomic data. BMC Bioinformatics, 19(7), 198.
Frank, E., Hall, M., Trigg, L., Holmes, G., & Witten, I. H. (2004). Data mining in bioinformatics using Weka. Bioinformatics, 20(15), 2479– 2481. https://doi.org/10.1093/bioinformatics/bth261
Free Software Foundation, I. (2016). GNU R. Retrieved from http://directory.fsf.org/wiki/R#tab=Overview
Gauthier, J., Vincent, A. T., Charette, S. J., & Derome, N. (2018). A brief history of bioinformatics. Briefings in Bioinformatics, (February), 1–16. https://doi.org/10.1093/bib/bby063
Goodfellow, I., Pouget-Abadie, J., Mirza, M., Xu, B., Warde-Farley, D., Ozair, S., … Bengio, Y. (2014). Generative adversarial nets. In Advances in neural information processing systems (pp. 2672–2680).
Guimaraes, G. L., Sanchez-Lengeling, B., Outeiral, C., Farias, P. L. C., & AspuruGuzik, A. (2017). Objective-reinforced generative adversarial networks (ORGAN) for sequence generation models. ArXiv Preprint ArXiv:1705.10843
Gupta, A., & Zou, J. (2018). Feedback GAN (FBGAN) for DNA: A novel feedback-loop architecture for optimizing protein functions. ArXiv Preprint ArXiv:1804.01694.
Hall, M., Frank, E., Holmes, G., Pfahringer, B., Reutemann, P., & Witten, I. H. (2009). The WEKA data mining software. ACM SIGKDD Explorations, 11(1), 10–18. https://doi.org/10.%201145/1656274.1656278
Hornik, K., Buchta, C., & Zeileis, A. (2009). Opensource machine learning: R meets Weka. Computational Statistics, 24(2), 225–232.
James, G., Witten, D., Hastie, T., & Tibshirani, R. (2013). An introduction to statistical learning (Vol. 112). Springer.
Karatzoglou, A., Smola, A., Hornik, K., & Zeileis, A. (2004). kernlab-an S4 package for kernel methods in R. Journal of Statistical Software, 11(9), 1–20.
Kelley, D. R., Reshef, Y. A., Bileschi, M., Belanger, D., McLean, C. Y., & Snoek, J. (2018). Sequential regulatory activity prediction across chromosomes with convolutional neural networks. Genome Research, 28(5), 739–750.
Kuhn, M. (2008). Building predictive models in R using the caret package. Journal of Statistical Software, 28(5), 1–26.
Kumar, A., & Chrodia, N. (2016). Role of Bioinformatics in Biotechnology. Research and Review in BioSciences, 12(1), 293–317. https://doi.org/10.4018/978-1-5225-0610-2.ch011
Lavecchia, A. (2015). Machine-learning approaches in drug discovery: Methods and applications. Drug Discovery Today, 20(3), 318–331. https://doi.org/10.1016/j.drudis.2014.10.012
LeCun, Y., Bengio, Y., Hinton, G., Y., L., Y., B., & G., H. (2015). Deep learning. Nature, 521(7553), 436–444. https://doi.org/10.1038/nature14539
Liakos, K. G., Busato, P., Moshou, D., Pearson, S., & Bochtis, D. (2018). Machine learning in agriculture: A review. Sensors (Switzerland), 18(8), 1–29. https://doi.org/10.3390/s18082674
Liaw, A., & Wiener, M. (2002). Classification and regression by randomForest. R News, 2(3), 18–22
Libbrecht, M. W., & Noble, W. S. (2015). Machine learning applications in genetics and genomics. Nat Rev Genet, 16(6), 321–332. https://www.doi.org/%2010.1038/nrg3920
Libbrecht, M. W., & Noble, W. S. (2017). Machine learning in genetics and genomics. Nature Reviews Genetics, 16(6), 321–332. https://doi.org/10.1038/nrg3920.Machine
Mamoshina, P., Vieira, A., Putin, E., & Zhavoronkov, A. (2016a). Applications of Deep Learning in Biomedicine. Molecular Pharmaceutics, 13(5), 1445–1454. https://doi.org/10.1021/acs.molpharmaceut.5b00982
Mamoshina, P., Vieira, A., Putin, E., & Zhavoronkov, A. (2016b). Applications of Deep Learning in Biomedicine. Molecular Pharmaceutics, 13(5), 1445–1454. https://doi.org/10.1021/acs.molpharmaceut.5b00982
Mamoshina, P., Volosnikova, M., Ozerov, I. V., Putin, E., Skibina, E., Cortese, F., & Zhavoronkov, A. (2018). Machine learning on human muscle transcriptomic data for biomarker discovery and tissue-specific drug target identification. Frontiers in Genetics, 9(JUL), 1–10. https://doi.org/10.3389/fgene.2018.00242
Martinez, R., Pasquier, N., & Pasquier, C. (2008). GenMiner: Mining non-redundant association rules from integrated gene expression data and annotations. Bioinformatics, 24(22), 2643– 2644. https://doi.org/10.1093/bioinformatics/btn490
Mccombie, W. R., Mcpherson, J. D., & Mardis, E. R. (2019). Next-Generation Sequencing Technologies. https://doi.org/10.1101/cshperspect.a036798
Metzker, M. L. (2010). Sequencing technologies — the next generation. Nature Reviews Genetics, 11(1), 31–46. https://doi.org/10.1038/nrg2626
Meyer, D., Dimitriadou, E., Hornik, K., Weingessel, A., & Leisch, F. (2017). e1071: misc functions of the department of statistics, probability theory group (formerly: E1071), TU Wien. R package version 1.6-8.
Mierswa, I., Wurst, M., Klinkenberg, R., Scholz, M., & Euler, T. (2006). YALE: Rapid prototyping for complex data mining tasks. Proceedings of the ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, 2006, 935–940.
Min, S., Lee, B., & Yoon, S. (2017). Deep learning in bioinformatics. Briefings in Bioinformatics, 18(5), 851–869. https://doi.org/10.1093/bib/bbw068
Mohanty, S. P., Hughes, D. P., & Salathé, M. (2016). Using deep learning for image-based plant disease detection. Frontiers in Plant Science, 7, 1419
Morales, I. R., Cebrián, D. R., Fernandez-Blanco, E., & Sierra, A. P. (2016). Early warning in egg production curves from commercial hens: A SVM approach. Computers and Electronics in Agriculture, 121(03082), 169–179. https://doi.org/10.1016/j.compag.2015.12.009
Noble, W. S. (2006). What is a support vector machine? Nature Biotechnology, 24(12), 1565–1567.
Oyelade, J., Isewon, I., Oladipupo, F., Aromolaran, O., Uwoghiren, E., Ameh, F., … Adebiyi, E. (2016). Clustering Algorithms: Their Application to Gene Expression Data. Bioinformatics and Biology Insights, 10, BBI. S38316. https://doi.org/10.4137/BBI.S38316
Pan, X., Rijnbeek, P., Yan, J., & Shen, H.-B. (2018). Prediction of RNA-protein sequence and structure binding preferences using deep convolutional and recurrent neural networks. BMC Genomics, 19(1), 511.
Park, S., Min, S., Choi, H., & Yoon, S. (2016). deepMiRGene: Deep neural network based precursor microrna prediction. ArXiv Preprint ArXiv:1605.00017
Park, Y., & Kellis, M. (2015). Deep learning for regulatory genomics. Nature Biotechnology, 33(8), 825–826. https://doi.org/10.1038/nbt.3313
Paszke, A., Gross, S., Chintala, S., Chanan, G., Yang, E., DeVito, Z., … Lerer, A. (2017). Automatic differentiation in pytorch
Patil, A. P., & Deka, P. C. (2016). An extreme learning machine approach for modeling evapotranspiration using extrinsic inputs. Computers and Electronics in Agriculture. https://doi.org/10.1016/j.compag.2016.01.016
Pedregosa, F., Michel, V., Grisel O., Blondel, M., Prettenhofer, P., Weiss, R., … Duchesnay E., Fré. (2011). Scikit-learn: Machine Learning in Python Gaël Varoquaux Bertrand Thirion Vincent Dubourg Alexandre Passos Pedregosa, Varoquaux, Gramfort et al. Matthieu Perrot. Journal of Machine Learning Research, 12, 2825–2830. Recuperado de http://scikit-learn.sourceforge.net
Peres-Neto, P. R., Jackson, D. A., & Somers, K. M. (2005). How many principal components? stopping rules for determining the number of non-trivial axes revisited. Computational Statistics and Data Analysis, 49(4), 974–997. https://doi.org/10.1016/j.csda.2004.06.015
Rhee, S., Seo, S., & Kim, S. (2017). Hybrid approach of relation network and localized graph convolutional filtering for breast cancer subtype classification. ArXiv Preprint ArXiv:1711.05859.
Ringnér, M. (2008). What is principal component analysis? Nature Biotechnology, 26(3), 303.
Rouillard, A. D., Hurle, M. R., & Agarwal, P. (2018). Systematic interrogation of diverse Omic data reveals interpretable, robust, and generalizable transcriptomic features of clinically successful therapeutic targets. PLoS Computational Biology, 14(5), 1–28. https://doi.org/10.1371/journal.pcbi.1006142
Scholz, M., Kaplan, F., Guy, C. L., Kopka, J., & Selbig, J. (2005). Non-linear PCA: a missing data approach. Bioinformatics, 21(20), 3887–3895.
Searls, D. B. (2010). The roots of bioinformatics. PLoS Computational Biology, 6(6), 1–7. https://doi.org/10.1371/journal.pcbi.1000809
Seide, F., & Agarwal, A. (2016). CNTK: Microsoft’s open-source deep-learning toolkit. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining (p. 2135). ACM.
Sheehan, S., & Song, Y. S. (2016). Deep learning for population genetic inference. PLoS Computational Biology, 12(3), e1004845
SINGH, V., SINGH, A., CHAND, R., & KUSHWAHA, C. (2011). Role of Bioinformatics in Agriculture and Sustainable Development. International Journal of Bioinformatics Research, 3(2), 221–226. https://doi.org/10.9735/0975-3087.3.2.221-226
Song, X., Zhang, G., Liu, F., Li, D., Zhao, Y., & Yang, J. (2016). Modeling spatio-temporal distribution of soil moisture by deep learning-based cellular automata model. Journal of Arid Land, 8(5), 734–748.
Tan, J., Doing, G., Lewis, K. A., Price, C. E., Chen, K. M., Cady, K. C., … Greene, C. S. (2017). Unsupervised extraction of stable expression signatures from public compendia with an ensemble of neural networks. Cell Systems, 5(1), 63–71
Tan, J., Hammond, J. H., Hogan, D. A., & Greene, C. S. (2016). ADAGE-based integration of publicly available Pseudomonas aeruginosa gene expression data with denoising autoencoders illuminates microbe-host interactions. MSystems, 1(1), e00025-150
Thermes, C. (2014). Ten years of next-generation sequencing technology. Trends in Genetics : TIG, 30(9), 418–426. https://doi.org/10.1016/j.tig.2014.07.001
Tiwari, A., & Sekhar, A. K. T. (2007). Workflow based framework for life science informatics. Computational Biology and Chemistry. https://doi.org/10.1016/j.compbiolchem.2007.08.009
Van Gerven, M., & Bohte, S. (2017). Artificial neural networks as models of neural information processing. Frontiers in Computational Neuroscience, 11, 114.
Wainberg, M., Merico, D., Delong, A., & Frey, B. J. (2018). Deep learning in biomedicine. Nature Biotechnology, 36(9), 829–838. https://doi.org/10.1038/nbt.4233
Wang, M., Tai, C., E, W., & Wei, L. (2018). DeFine: deep convolutional neural networks accurately quantify intensities of transcription factor-DNA binding and facilitate evaluation of functional non-coding variants. Nucleic Acids Research, 46(11), e69–e69
Werli, S. (2016). scikit-learn: Classification Algorithms on Iris Dataset - Brain Scribble. Retrieved September 21, 2019, from http://stephanie-w.github.io/brainscribble/classification-algorithms-on-iris-dataset.htm
Witten, I. H., Frank, E., & Hall, M. a. (2011a). Data Mining: Practical Machine Learning Tools and Techniques, Third Edition. Annals of Physics (Vol. 54). https://doi.org/10.1002/1521-3773 (20010316)40:6<9823::AID-ANIE9823 >3.3.CO;2-C
Witten, I. H., Frank, E., & Hall, M. A. (2011b). Data Mining Practical Machine Learning Tools and Techniques (3ra ed.). Burlington, MA: Morgan Kaufmann.
Yadav, B., Ch, S., Mathur, S., & Adamowski, J. (2016). Estimation of in-situ bioremediation system cost using a hybrid Extreme Learning Machine (ELM)-particle swarm optimization approach. Journal of Hydrology. https://doi.org/10.1016/j.jhydrol.2016.10.013
Zhou, J., Theesfeld, C. L., Yao, K., Chen, K. M., Wong, A. K., & Troyanskaya, O. G. (2018). Deep learning sequence-based ab initio prediction of variant effects on expression and disease risk. Nature Genetics, 50(8), 1171.
Zou, Z., Yang, L., Wang, D., Huang, Q., Mo, Y., & Xie, G. (2016). Gene Structures , Evolution and Transcriptional Profiling of the WRKY Gene Family in Castor Bean ( Ricinus communis L.), 1–23. https://doi.org/10.1371/journal.pone.0148243
