Ciencia, Ambiente y Clima, Vol. 2, No. 2, julio-diciembre, 2019 • ISSN: 2636-2317 (impreso) | 2636-2333 (en línea) • Sitio web: https://revistas.intec.edu.do/
UMPOLUNG IN REACTIONS CATALYZED BY THIAMINE PYROPHOSPHATE DEPENDENT ENZYMES
Polaridad inversa en reacciones catalizadas por tiamina Enzimas dependientes de pirofosfato
*Instituto Tecnológico de Santo Domingo (INTEC), Área de Ciencias Básicas y Ambientales, Av. de Los Próceres 49, Santo Domingo, República Dominicana Corresponding author: Carlos J. Boluda. Correo-e: carlos.boluda@intec.edu.do
**Instituto Tecnológico de Santo Domingo (INTEC), Área de Ciencias Básicas y Ambientales. Correo-e: carolinajuncam yahoo.com
***Instituto Tecnológico de Santo Domingo (INTEC), Área de Ciencias Básicas y Ambientales. Correo-e: emilysotoh@hotmail.com
****Instituto Tecnológico de Santo Domingo (INTEC), Área de Ciencias Básicas y Ambientales. Correo-e: darahsotoh@hotmail.com
*****Instituto Tecnológico de Santo Domingo (INTEC), Área de Ciencias Básicas y Ambientales. Correo-e: annyggps@gmail.com
Recibido: ● Aprobado:

Cómo citar: J. Boluda, C., Juncá, C., Soto, E., de la Cruz, D., & Peña, A. (2019). Umpolung in reactions catalyzed by thiamine pyrophosphate dependent enzymes. Ciencia, Ambiente y Clima, 2(2), 27-42. Doi: https://doi.org/10.22206/cac.2019.v2i2.pp27-42
Resumen
El intercambio temporal del carácter electrofílico/nucleofílico de un átomo mediante manipulación química, es conocido con el vocablo alemán de umpolung. Esta inversión de polaridad permite explorar nuevas posibilidades sintéticas que no pueden llevarse a cabo partiendo de la reactividad normal de los grupos funcionales. Se trata de una herramienta sintética útil, que también es utilizada por ciertas enzimas en determinadas reacciones bioquímicas que tienen lugar en las células. Las enzimas dependientes del pirofosfato de tiamina, como la piruvato descarboxilasa, la piruvato deshidrogenasa, la α-cetoglutarato deshidrogenasa, la deshidrogenasa de α-cetoácidos de cadena ramificada y la transcetolasa, proporcionan ejemplos claros de umpolung en reacciones celulares. En esta revisión, tras una discusión sobre el significado del término umpolung encontrado en la literatura química, se analizan los mecanismos de reacción y el significado bioquímico de las transformaciones llevadas a cabo por las enzimas mencionadas.
Palabras clave:
Abstract
The temporal exchange of the electrophilic/nucleophilic character of an atom by chemical manipulation is known in organic chemistry as umpolung. This inversion of polarity allows the exploration of new synthetic possibilities which cannot be achieved from the normal reactivity of functional groups. It is not only a useful synthetic tool, but it is also utilized by certain enzymes during biochemical reactions in living organisms. Thiamine pyrophosphate dependent enzymes, such as pyruvate decarboxylase, pyruvate dehydrogenase, α-ketoglutarate dehydrogenase, branched-chain α-keto acids dehydrogenase and transketolase, provide clear examples of umpolung in cellular reactions that fulfill different purposes. In this review, after a discussion regarding the meaning of the term umpolung found in the chemical literature, the reaction mechanisms and the biochemical meaning of transformations carried out by the aforementioned enzymes are analyzed.
Keywords:
1. Introduction
Multiple reactions in cellular metabolism consist in C-C bond formation or breakage. During glycolysis, the retro-aldol reaction catalyzed by aldolase, in which the fructose-1,6-diphosphate splits in two phosphorylated trioses, represents an example of C-C bond cleavage.
Usually, the linkage of carbons occurs between atoms with opposite polarities (electrophile/nucleophile). This can be observed in reactions such as the Claisen condensation between acetyl-CoA and oxaloacetate (OAA), catalyzed by citrate synthase during the Krebs cycle. In this reaction, both the carbonyl group and the enolate keep their typical reactivity, them being electron acceptor and electron donor, respectively (Nelson, Cox & Lehninger, 2013). Figure 1 shows the enolate of acetyl-CoA (1) reacting with the carbonyl group of the OAA (2) to form a new C-C bond in the citrate molecule (3).
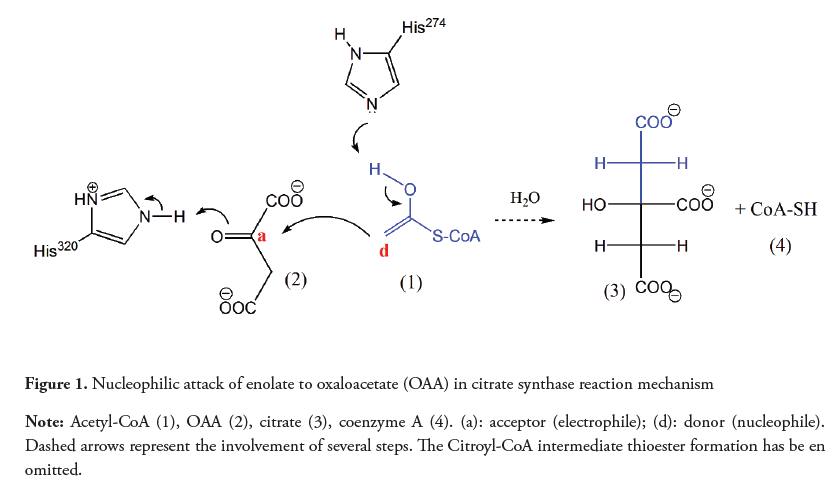
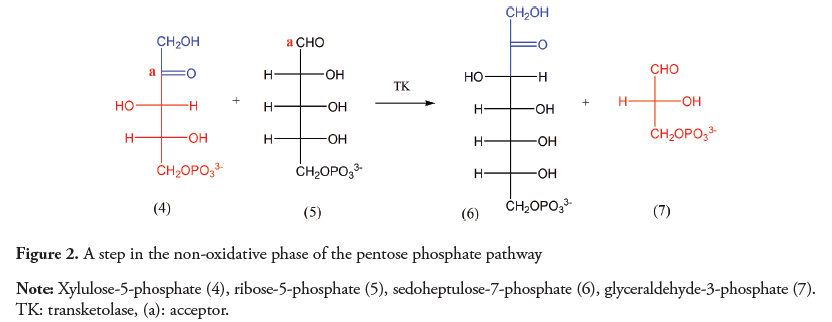
The formation of C-C bonds between carbons sharing the same polarity is also possible. Nevertheless, it requires the reversal of the electron donor/acceptor character of one of the atoms, a phenomenon known as umpolung. The carbonyl group is intrinsically an acceptor, and therefore reacts with electron donors and not with another electron deficient carbons. Examples of reactions between carbonyl groups to form a C-C bond are found in metabolic pathways such as the Calvin cycle and in the non-oxidative phase of the pentose phosphate. In the latter, as shown in Figure 2, a C-2 fragment must be transferred from a ketopentose (4) to an aldopentose (5), resulting in a ketoheptose (6) and a triose (7). This reaction is catalyzed by the transketolase, a thiamine pyrophosphate (TPP) dependent enzyme which belongs to a diverse group of biocatalysts involved in the rupture and/ or formation of C-C bonds (Mosbacher, Mueller & Schulz, 2005).
The objective of this review is to illustrate how the inversion of the original polarity of the carbonyl group in certain cellular metabolites takes place inside the active site of TPP-dependent enzymes. Additionally, the biochemical meaning of these inversions is analyzed.
2. Umpolung
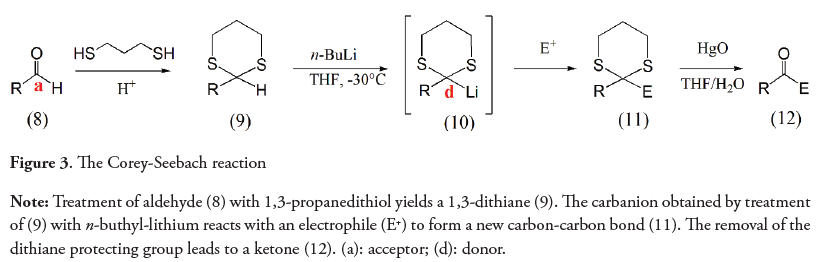
Umpolung is a German word, used since the mid-twentieth century (Wittig, Davis & Koenig, 1951; Seebach & Corey, 1975), that refers to any process by which the nucleophilic (electron donor) or electrophilic (electron acceptor) character of an atom in a molecule is exchanged (Seebach, 1979). This inversion of the atoms inherent polarity allows the carrying out of reactions that are not possible from the original reactivity of its functional group (Eymur, Göllü & Tanyeli, 2013). The Corey-Seebach reaction is a classic example of umpolung in which the inversion of the normal reactivity of a carbonyl group (electrophile) is produced. This allows the carbonyl carbon to react with electrophiles as an acyl anion, exhibiting the opposite chemical behavior of its natural reactivity and being very useful in organic synthesis (Corey & Seebach, 1965).
As shown in Figure 3, in the Corey-Seebach reaction the treatment of 1,3-dithianes (9) with n-butyl-lithium leads to a carbanion (10) with an excellent nucleophilic reactivity (Corey & Seebach, 1965). The carbanions thus formed can be used in multiple reactions in which a new bond is formed between two originally electrophilic carbons.
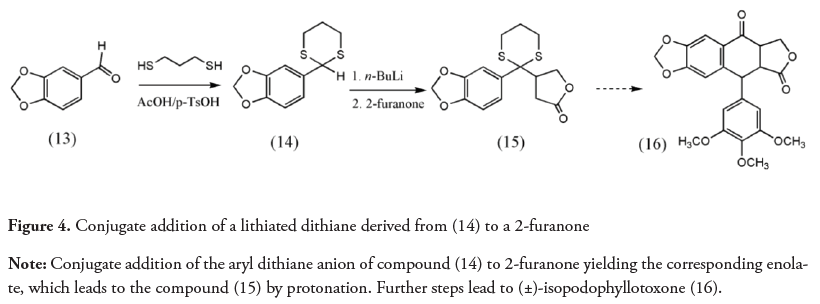
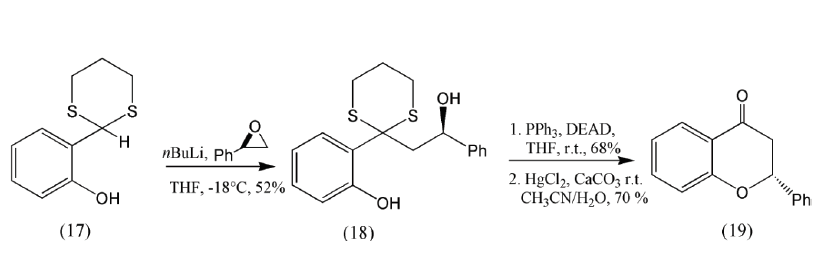
The Corey-Seebach reaction has been widely used in natural product synthesis (Yus, Nájera & Foubelo, 2003). Figure 4 shows the conjugate addition of a lithiated dithiane derived from (14) to 2-furanone; this sequence of reactions has been employed in (±)-podorhizol, (±)-isopodophyllotoxone (Ziegler & Schwartz,1978) and in arylnaphtalene lignans synthesis (González, Pérez & Trujillo, 1978; Boluda, López, Pérez & Trujillo, 2005). Figure 5 shows the strategy used by Noda and Watanabe in the enantiospecific synthesis of the (R)- and (S)-enantiomers of a flavanone. The nucleophilic addition of the carbanion of a dithiane (17) to a chiral epoxide forms a diol (18) that under Mitsunobu cyclization yields the synthetic target (19) (Noda & Watanabe, 2002). The dithianes have also been used to obtain more complex natural products as spongistatins (Smith et al., 2001).
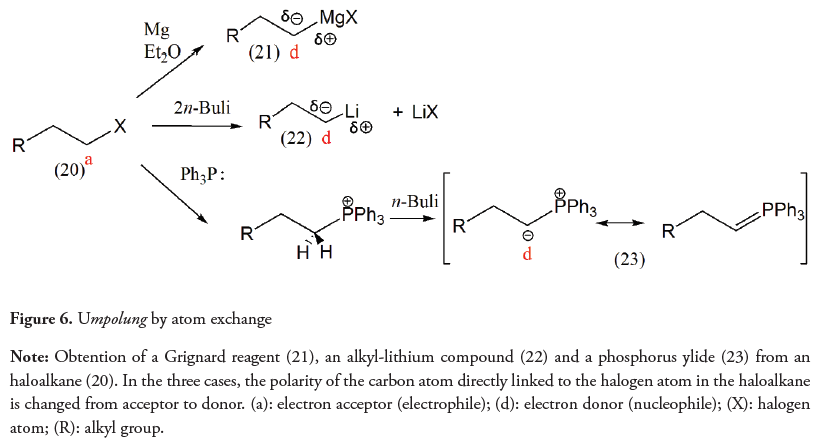
Organometallic reagents such as Grignard reagents or alkyl-lithium and phosphorus ylides shown in Figure 6 provide clear examples of umpolung by atom exchange (Arndt, Kunde & Mahrwald, 2016).
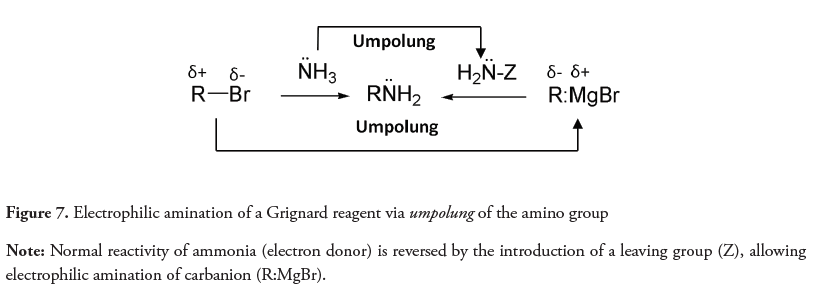
The umpolung is a synthetic tool which includes the reversion of the normal reactivity of amines (donors) by replacing one or more of their hydrogens with good leaving groups. This strategy provides another method of obtaining amines from organometallic compounds such as Grignard reagents (RMgBr), as an alternative to the alkylation of ammonia and primary amines, which takes advantage of the normal reactivity of the amines (Figure 7) (Erdik & Ay, 1989). Another example of amine umpolung utilizes N-haloamines and α-halonitroalkanes, as an acyl donor, for obtaining amides (Shen, Makley & Johnston, 2010).
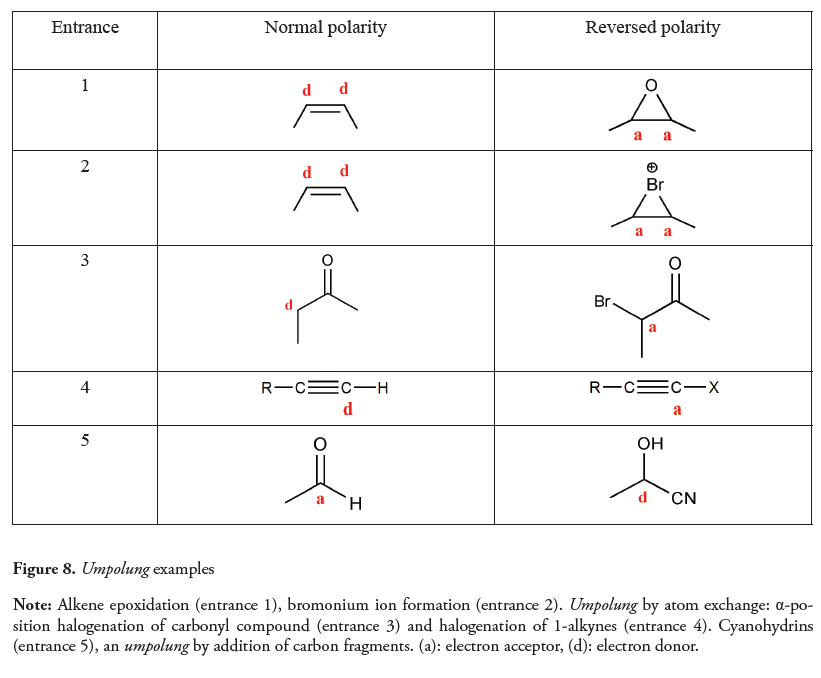
As shown in Figure 8, other umpolung examples include the inversion of the inherent reactivity of alkenes, umpolung by atom exchange and umpolung by addition of carbon fragments.
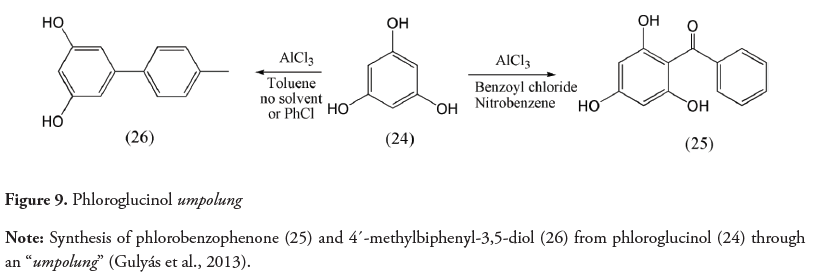
The term umpolung has also been used in a broader sense to indicate a change in the reactivity of a compound and not to refer to a particular atom. In 2010 Guérard, Sabot, Beaulieu, Giroux, & Canesi utilized the expression “Aromatic ring umpolung” to refer to the formation of a highly electrophilic phenoxenium ion which reacts with nucleophiles. These acceptor species were formed from phenols by treating them with an oxidizing agent such as diacetoxyiodobenzene (DIB) (Guérard et al., 2010). Additionally, in 2013 the inversion of the normal polarity in phloroglucinol (1,3,5-trihydroxybenzene) was reported. This compound is expected to behave as a nucleophile in an aromatic electrophilic substitution reaction (Figure 9) but reacts as an electrophile under certain conditions. Phloroglucinol (24) reacts as an electron donor under typical Friedel-Crafts reaction conditions with benzoyl chloride in nitrobenzene to form phlorobenzophenone (25), although with low yield. Nevertheless, when reacting with toluene and no solvent or with chlorobenzene, it behaves as an electrophile forming 4´-methylbiphenyl-3,5-diol (26) with an 89% yield. This “umpolung” example observed in phloroglucinol allows a simple way to obtain 5-aryl-substituted resorcinols (Gulyás, Boluda, Westermann & Wessjohann, 2013).
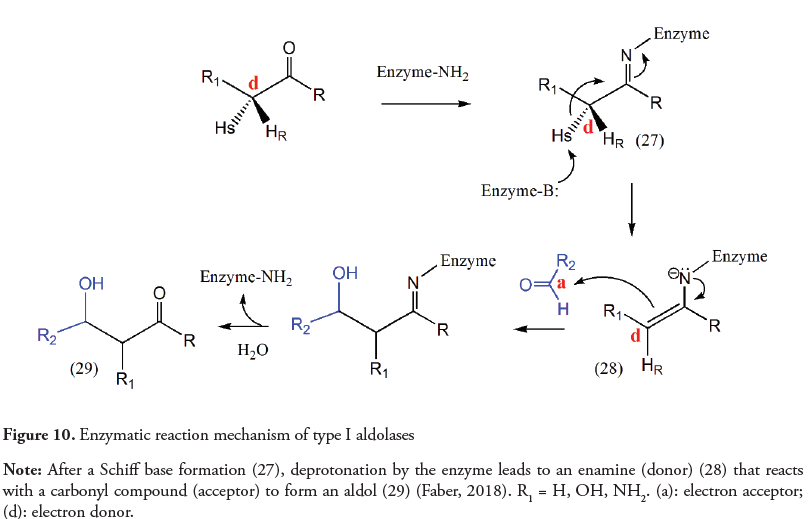
This umpolung lato sensu has also been used to describe the aldol reaction mechanism carried out by aldolases. This enzymatic transformation involves the formation of an enamine (aldolases type I) or an enolate (aldolases type II) in the active site of the enzyme. Then the donor performs a nucleophilic addition to an acceptor (Figure 10) (Faber, 2018). However, the abstraction of a hydrogen from the α-position by the enzyme should not be considered strictly an umpolung example, since that α-carbon is a donor (Arndt et al., 2016) and no inversion of polarity takes place; the enzyme only takes advantage of the normal polarity of the molecule, increasing its reactivity. Nevertheless, an umpolung stricto sensu, as defined by Seebach in 1979, can be observed in the mechanism of enzymatic action of TPP dependent enzymes.
3. Thiamine pyrophosphate (TPP)
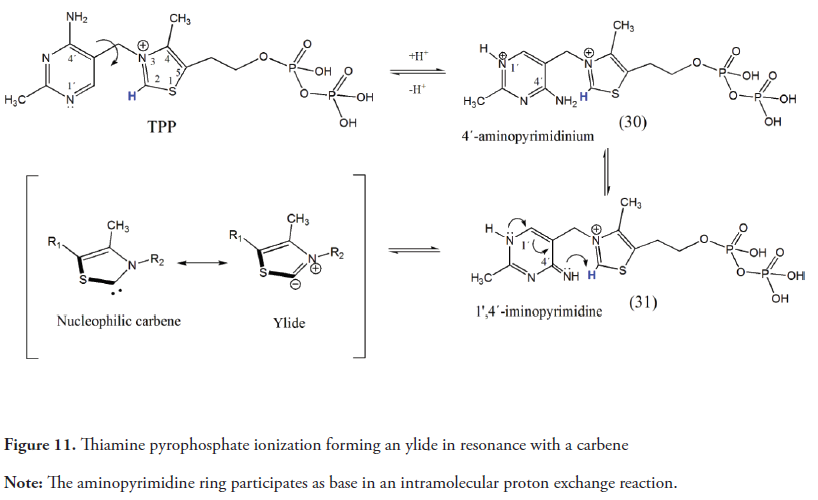
Thiamine pyrophosphate (TPP) (Figure 11) is a vitamin B1 derived coenzyme (Jong, Meng, Dent & Hekimi, 2004) used by a diverse collection of enzymes that catalyze the formation and rupture of carbon-carbon bonds adjacent to a carbonyl group. Additionally, the TPP is an allosteric regulator of enzymes in cellular metabolism and has a relevant role in the response to several environmental factors such as pathogens and oxidative stress (Tylicki, Lotowski, Siemieniuk & Ratkiewicz, 2018). According to the NCBI PubChem Database (2019), the chemical structure of TPP consists of a methylamino pyrimidine ring linked through a methylene bridge to a methylthiazolium ring. A pyrophosphate group is attached to its hydroxyethyl side chain (Figure 11). This methylthiazolium ring of TPP behaves as an electron sink during reactions and stabilizes what would otherwise be an unstable acyl carbanion intermediate in the form of an enamine (Agyei-Owusu & Leeper, 2009; Nelson et al., 2013).
As shown in Figure 11, the thiazolium ring proton (C-2) is slightly acidic (pKa≈18) and its deprotonation results in the formation of an ylide with a nucleophilic carbene as a resonance form, as proposed by Breslow in 1958. The aminopyrimidine ring participates actively in the TPP ionization (Figure 11). The deprotonated form of the imino tautomer (31) of 4 ‘-aminopyridinium (30) acts as an internal base accepting the proton of the thiazolium ring (Agyei-Owusu & Leeper, 2009; Balakrishnan et al., 2012).
The carbene stabilized by adjacent heteroatoms (Hanson, 1987; Prier & Arnold, 2015; Heidari, Howe & Kluger, 2016) reacts as a strong nucleophile, by addition with the carbonyl group of a metabolite (Broderick, 2001; Amyes & Richard, 2017).
In absence of the apoenzyme, TPP decarboxylates pyruvic acid at room temperature at pH 8.4 (Mizuhara, Tamura & Arata, 1951; Breslow, 1958). Therefore, promoting the non-enzymatic decarboxylation of α-keto acids seems to be an inherent ability of this coenzyme. Furthermore, other thiazolium compounds share this catalytic capacity and mimic the TPP enzymatic reactions (Breslow, 1958; Coopper, Ginos & Meister, 1983; Nemeria, Chakraborty, Balakrishnan & Jordan, 2009).
4. TPP dependent dehydrogenases: PDHC, KGDHC and BCKDHC
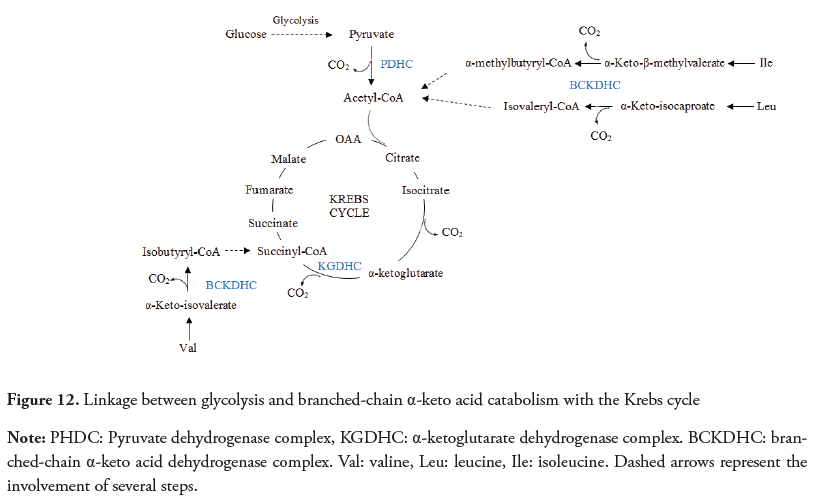
Pyruvate dehydrogenase complex (PDHC), α-ketoglutarate dehydrogenase complex (KGDHC), and branched-chain α-keto acid dehydrogenase complex (BCKDHC) are mitochondrial enzymatic complexes composed by multiple copies of three enzymes and five coenzymes. Their basic structure has been preserved during evolution and allows the transformation of their specific α-keto acids, into their corresponding thioesters through a decarboxylation reaction (Lonsdale, 2006; Nelson et al., 2013), while also catalyzing the reduction of nicotinamide adenine dinucleotide (NAD +) to NADH (Fries, Jung & Perham, 2003).
PDHC oxidizes pyruvate to acetyl coenzyme A (acetyl CoA), linking glycolysis and the Krebs cycle (Figure 12) (Tylicki et al., 2018); KGDHC catalyzes the oxidative decarboxylation of α-ketoglutarate to form succinyl-CoA in the tricarboxylic acid cycle; and the BCKDHC produces the initial and irreversible step in the catabolism of branchedchain amino acids (valine, leucine and isoleucine) to form the respective thioesters with coenzyme A (Wynn, Daviel, Mengl & Chuang, 1996). After the transamination of these amino acids to their corresponding α-keto acids, this enzymatic complex is responsible for catalyzing the oxidative decarboxylation of the branched-chain α-keto acids (Figure 12) (Johnson, Yang & Patel, 2000; Wynn et al., 2004).
As the PDH and KGDH complexes, the BCKDHC consists of three catalytic components: α-keto acid dehydrogenase (subunit E1), dihydrolipoyl transacylase (subunit E2) and dihydrolipoyl dehydrogenase (subunit E3) (Ævarsson et al., 2000; Wynn et al., 2004). The complex formed by these enzymes needs five cofactors to perform its function: FAD (flavin adenine dinucleotide), NAD+ (nicotinamide adenine dinucleotide), TPP (thiamine diphosphate), lipoic acid, and coenzyme A (Johnson et al., 2000).
The E1 component itself is a tetramer composed of two subunits (E1 alpha and E1 beta) with TPP as required coenzyme for the decarboxylation of branched-chain α-keto acids (Heffelfinger, Sewell & Danner, 1983; Li, Huo, Pulley & Liu, 2012).
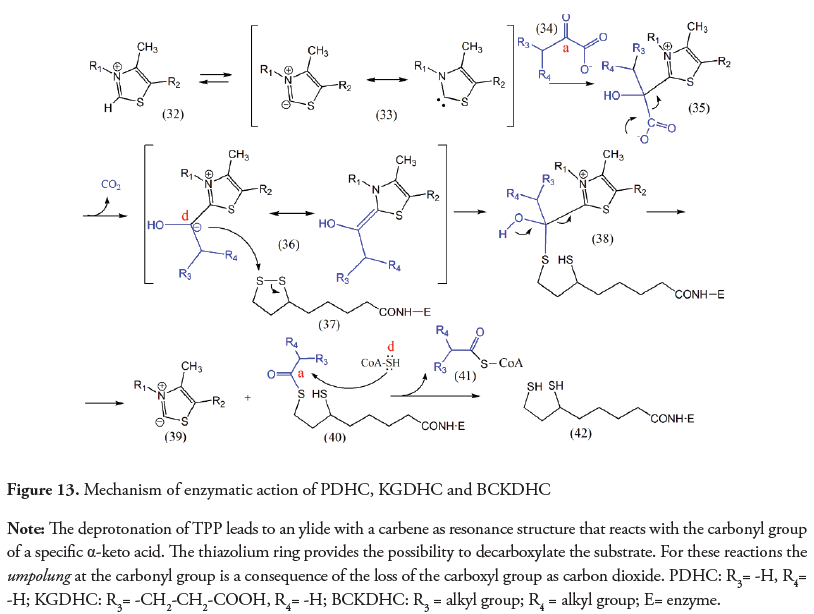
This conversion, illustrated in Figure 13, involves an umpolung. When the TPP binds to the enzyme, the deprotonation of the thiazolium ring at C2 (32) creates a nucleophilic ylide resonant with a carbene (33) (Tittmann et al., 2003). Once the branched-chain α-keto acid (34) reacts with the ylide/carbene, it forms a tetrahedral covalent intermediate (35). The subsequent decarboxylation of this substrate results in a stabilized carbanion (36) (Fiedler et al., 2002; Kluger, 1987). The carbanion is then transferred to the lipoamide (37), restoring the original polarity of the carbon atom (acceptor) (40). The addition-elimination sequence with coenzyme A acting as nucleophile (transthioesterification) forms a new thioester (acyl-CoA), releasing a branched acyl-CoA (41) and the dihydrolipoamide (42).
5. Pyruvate decarboxylase
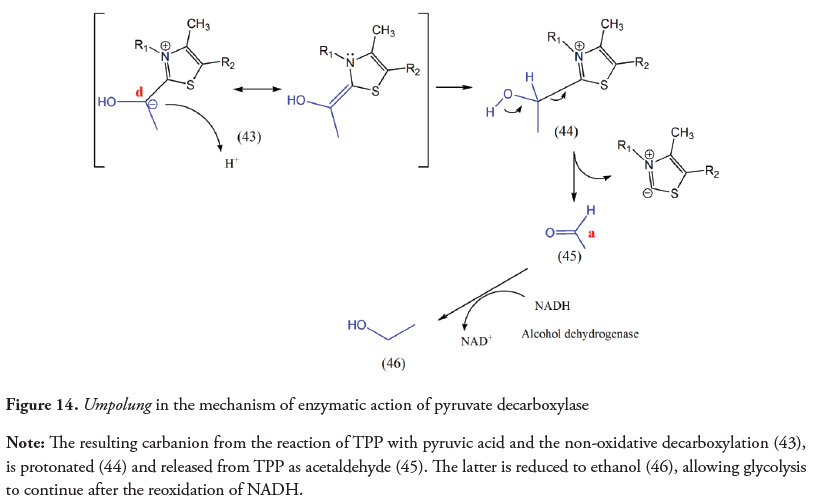
Yeast and all organisms that ferment glucose to ethanol use the TPP-dependent enzyme complex pyruvate decarboxylase (PDC) to catalyze the non-oxidative decarboxylation of pyruvate and produce acetaldehyde. This is then converted to ethanol by alcohol dehydrogenase (Hanson, 1987; Nelson et al., 2013) reoxidizing NADH to sustain glycolysis (Nelson et al., 2013; Tylicki et al., 2018). Even though the mechanism of PDC is similar to that of PDH, KGDH and BCKDH, the enamine produced is protonated at the C2 α-position, releasing acetaldehyde (Eram & Ma, 2013).
6. Transketolase
Transketolase (TK), also called glycolaldehyde transferase, is an enzyme that uses thiamine pyrophosphate (TPP) as its coenzyme and a divalent metal cation (Mg2+ or Ca2+ ) in order to perform the cleavage of carbon-carbon bonds and transfer a dihydroxyethyl unit from ketose donors to aldose acceptors (Schenk, Duggleby & Nixon, 1998; Fullam, Pojer, Bergfors, Jones & Cole, 2012). From an organic chemistry perspective, the latter seems not to be a trivial task, but a challenge for cellular chemistry due to its occurrence in an aqueous media and with phosphorylated and non-phosphorylated sugars as substrate (Miyamoto & Ohta, 2007).
The TK is involved in the Calvin cycle and non-oxidative phase of the pentose phosphate pathway. In both cases, the enzyme transfers two carbon fragments from ketosugars such as xylulose-5-phosphate or fructose-6-phosphate, to aldoses like ribose-5-phosphate or glyceraldehyde-3-phosphate in order to assemble precursors of other molecules in each route (Figure 15) (Schenk, Duggleby & Nixon, 1998; Miyamoto & Ohta, 2007; Lonsdale, 2006).
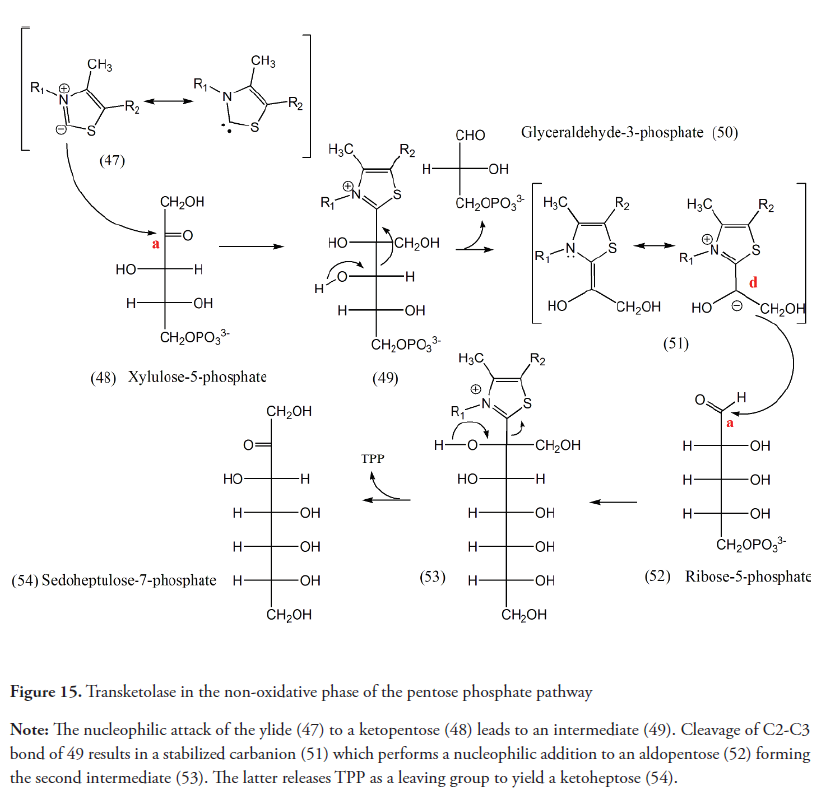
The C-C bond formation catalyzed by TK requires an umpolung stricto sensu. The nucleophilic attack from ionized TPP to a carbonyl group of the ketosugar produces a tetrahedral intermediate. The polarity inversion occurs because of the cleavage of this intermediate releasing an aldehyde and the resulting stabilized carbanion which is then transferred to the aldose. After the cleavage, a ketone and the TPP are released (Schneider & Lindqvist, 1993).
7. Conclusions
Although the scope of the term umpolung is usually restricted to organic chemistry, it is certainly a tool available to living organisms. Inside the cell, some chemical reactions require the formation of an anomalous synthon, an acyl carbanion (Agyei-Owusu & Leeper 2009). Due to the inability of the 20 amino acid residues normally found in enzymes to stabilize this intermediate, the coenzyme TPP has evolved to assume this role (Hanson, 1987). The mechanism of the TPP dependent enzymes analyzed provide a clear example of umpolung stricto sensu in nature, although with very different biochemical meanings.
In the case of pyruvate decarboxylase (PDC), the umpolung at C2 appears as a result of the non-oxidative decarboxylation and it seems not to obey a biosynthetic strategy, as the masked acyl anion (enamine) is protonated yielding acetaldehyde. The latter is reduced to ethanol by NADH providing the required NAD+ to continue glycolysis.
Nevertheless, in other enzymes that carry out a decarboxylation reaction of an α-keto acid, such as pyruvate dehydrogenase complex (PDHC) or the branched-chain amino acid dehydrogenase complex (BCKDHC), the umpolung is produced once more as a consequence of the loss of carbon dioxide, but it is used to obtain an acyl-CoA and NADH. As shown in Figure 13, once the decarboxylation takes place, the normal reactivity of the carbonyl group (acceptor) is restored by transferring the carbanion to the lipoyl moiety forming a thioester. The recovering of the normal polarity of the carbonyl group in this intermediate is necessary to react with coenzyme A and to yield an acyl-CoA.
Unlike the cases described above, in reactions catalyzed by transketolase, the inversion of the normal reactivity of the carbonyl group of a monosaccharide, consequence of the rupture of a C-C bond in the sugar skeleton, is utilized to perform a new bond between two originally electrophilic carbons. This allows the rearrangement reactions in the non-oxidative phase of the pentose phosphate pathway and the Calvin cycle.
The fact that the TPP has catalytic activity per se, catalyzing the decarboxylation of α-keto acids, suggests that this might be the primordial function of this coenzyme in the cells. Later on, during evolution, the ability of the coenzyme to generate masked and highly reactive acyl anions would be opportunistically used to achieve more complex catalytic challenges, as forming bonds between two originally electrophilic carbons, as in transketolase catalyzed reactions. Performing this umpolung in aqueous medium had to be a major problem, since the carbanion could be easily protonated by water.
Referencias
Arndt, A., Kunde, T. & Mahrwald, R., (2016). Logic of organic chemistry. ChemTexts: The textbook journal of chemistry, 2(1), 1-3. doi: http://doi.org/10.1007/s40828-015-0020-2
Avarsson, A. C., Chuang, J. L., Wynn, R. M., Turley, S., Chuang, D. T. & Hol, W. G. (2000). Crystal structure of human branched-chain α-keto acid dehydrogenase and the molecular basis of multienzyme complex deficiency in maple syrup urine disease. Structure, 8(3), 277-291. doi: http://doi.org/10.1016/s0969-2126(00)00105-2
Agyei-Owusu, K. & Leeper, F. J. (2009). Thiamin diphosphate in biological chemistry: analogues of thiamin diphosphate in studies of enzymes and riboswitches. FEBS Journal, 276(11), 2905-2916. doi: http://doi.org/10.1111/j.1742-%204658.2009.07018.x
Amyes, T. L. & Richard, J. P. (2017). Substituent Effects on Carbon Acidity in Aqueous Solution and at Enzyme Active Sites. Synlett: accounts and rapid communications in synthetic organic chemistry, 28(12), 2407-2421. doi: http://doi.org/10.1055/s-0036-1588778
Balakrishnan, A., Gao, Y., Moorjani, P., Nemeria, N.S., Tittmann, K. & Jordan, F. (2012). Bifunctionality of the Thiamin Diphosphate Cofactor: Assignment of Tautomeric/Ionization States of the 4′-Aminopyrimidine Ring When Various Intermediates Occupy the Active Sites during the Catalysis of Yeast Pyruvate Decarboxylase. Journal of the American Chemical Society, 134(8), 3873-3885. doi: http://doi.org/10.1021/ja211139c
Boluda, C. J., López, H., Pérez, J. A. & Trujillo, J. M. (2005). First Total Synthesis of Justicidone, a p-Quinone-Lignan Derivative from Justicia hyssopifolia. Chemical and Pharmaceutical Bulletin, 53(8), 930-933. doi: http://doi.org/10.1248/cpb.53.930
Breslow, R. (1958). On the Mechanism of Thiamine Action. IV. Evidence from Studies on Model Systems. Journal of the American Chemical Society, 80(14), 3719-3726. doi: http://doi.org/10.1021/%20ja01547a064
Broderick, J. B. (2001). Coenzymes and Cofactors. Encyclopedia of Life Sciences, 1-11, doi: http://doi.org/10.1038/npg.els.0000631
Cooper, A. J. L., Ginos, J. Z. & Meister, A. (1983). Synthesis and Properties of the α-keto acids. Chemical Reviews, 83(3), 321-358, doi: http://doi.org/10.1021/cr00055a004
Corey, E. J. & Seebach, D. (1965). Carbanions of 1,3-Dithianes. Reagent for C-C Bond Formation by Nucleophilic Displacement and Carbonyl Addition. Angewandte Chemie International Edition in English, 4(12), 1075- 1077. doi: http://doi.org/10.1002/anie.196510752
Eram, M. S. & Ma, K. (2013). Decarboxylation of pyruvate to acetaldehyde for ethanol production by hyperthermophiles. Biomolecules, 3(3), 578-596. doi: http://doi.org/10.3390/biom3030578
Erdik, E. & Ay, M. (1989). Electrophilic Amination of Carbanions. Chemical Reviews, 89(8), 1947- 1980, doi: http://doi.org/10.1021/cr00098a014
Eymur, S., Göllü, M. & Tanyeli, C. (2013). Umpolung strategy: Advances in catalytic C-C bond formations. Turkish Journal of Chemistry, 37(4), 586-609. doi: http://doi.org/10.3906/kim-1303-85.
Fullam, E., Pojer, F., Bergfors, T., Jones, T. A. & Cole, S. T. (2012). Structure and function of the transketolase from Mycobacterium tuberculosis and comparison with the human enzyme. Open Biology, 2(1), 110026-110038. doi: http://doi.org/10.1098/rsob.110026
Faber, K. (2018). Biotransformations in Organic Chemistry: A Textbook. (pp. 205-206). (7th ed). Berlin, Alemania: Springer.
Fiedler, E., Thorell, S., Sandalova, T., Golbik, R., König, S. & Schneider, G. (2002). Snapshot of a key intermediate in enzymatic thiamine catalysis: Crystal structure of the α-carbanion of (α, β-dihydroxyethyl)-thiamin diphosphate in the active site of transketolase from Saccharomyces cerevisiae. Proceedings of the National Academy of Sciences, 99(2), 591-595. doi: http://doi.org/10.1073/pnas.022510999
Fries, M., Jung, H. & Perham, R. N. (2003). Reaction Mechanism of the Heterotetrameric (α2 β2 ) E1 Component of 2-Oxo Acid Dehydrogenase Multienzyme Complexes. Biochemistry, 42(23), 6996-7002. doi: http://doi.org/10.1021/bi027397z
González, A. G., Pérez, J. P. & Trujillo, J. M. (1978). Synthesis of two arylnaphthalene lignans. Tetrahedron, 34, 1011-1013. doi: http://doi.org/10.1016/0040-4020(78)88156-3
Guérard, K. C., Sabot, C., Beaulieu, M. A., Giroux, M. A. & Canesi, S. (2010). ‘Aromatic ring umpolung’, a rapid access to the main core of several natural products. Tetrahedron, 66(31), 5893- 5901. doi: http://doi.org/10.1016/j.tet.2010.03.09
Gulyás-Fekete, G., Boluda, C. J., Westermann, B. & Wessjohann, L. A. (2013). Anti-FriedelCrafts-Type substitution To Form Biaryl Linkages. Synthesis, 45(21), 3038-3043. doi: http://doi.org/10.1055/s-0033-1339682
Hanson, R. W. (1987). Decarboxylation of α-keto acids. Journal of Chemical Education, 64(7), 591-595. doi: http://doi.org/10.1021/ed064p591
Heidari, Y., Howe, G. W. & Kluger, R. (2016). The reactivity of lactyl-oxythiamine implies the role of the amino-pyrimidine in thiamin catalyzed decarboxylation. Bioorganic Chemistry, 69, 153- 158. doi: http://doi.org/10.1016/j.bioorg.2016.10.008
Heffelfinger, S. C., Sewell, E. T. & Danner, D. J. (1983). Identification of Specific Subunits of Highly Purified Bovine Liver Branched-Chain Ketoacid Dehydrogenase. Biochemistry, 22(24), 5519-5522. doi: http://doi.org/10.1021/bi00293a011
Johnson, M. T., Yang, H. S. & Patel, M. S. (2000). Targeting E3 Component of α-Keto Acid Dehydrogenase Complexes. Methods in Enzymology, 324, 465-476. Amsterdam: Elsevier. doi: http://doi.org/10.1016/s0076-6879(00)24254-7
Jong, L., Meng, Y., Dent, J. & Hekimi, S. (2004). Thiamine Pyrophosphate Biosynthesis and Transportin the Nematode Caenorhabditis elegans. Genetics Society of America, 168(2), 845- 854. doi: http://doi.org/10.1534/genetics.104.028605
Kluger, R. (1987). Thiamin diphosphate: A mechanistic update on enzymic and nonenzymic catalysis of decarboxylation. Chemical Reviews, 87(5), 863-876. doi: http://doi.org/10.1021/cr00081a001
Li, T., Huo, L., Pulley, C. & Liu, A. (2012). Decarboxylation mechanisms in biological system. Bioorganic Chemistry, 43, 2-14. doi: http://doi.org/10.1016/j.bioorg.2012.03.001
Lonsdale, D. (2006). A review of the biochemistry, metabolism and clinical benefits of thiamin(e) and its Derivatives. Evidence-Based Complementary and Alternative Medicine, 3(1), 49-59. doi: http://doi.org/10.1093/ecam/nek009
Miyamoto, K. & Ohta, H. (2007). Enzymatic decarboxylation of synthetic compounds. Future Directions in Biocatalysis, 305-343. doi: http://doi.org/10.1016/B978-044453059-2/50013-3
Mizuhara, S., Tamura, R. & Arata, H. (1951). On the Mechanism of Thiamine Action. II. Proceedings of the Japan Academy, 27(6), 302-308.
Mosbacher, T. G., Mueller, M. & Schulz, G. E. (2005). Structure and mechanism of the ThDPdependent benzaldehyde lyase from Pseudomonas fluorescens. FEBS Journal, 272(23), 6067-6076. doi: http://doi.org/10.1111/j.1742-4658.2005.04998.x
National Center for Biotechnology Information. PubChem Database. (2019, September 9). Thiamine diphosphate, CID=1132. Retrieved from https://pubchem.ncbi.nlm.nih.gov/compound/Thiamine-diphosphate
Nelson, D., Cox, M. & Lehninger, A. (2013). Lehninger Principles of Biochemistry, (513; 565; 605; 635), (6 ed). New York, USA: W.H. Freeman.
Nemeria, N. S., Chakraborty, S., Balakrishnan, A. & Jordan, F. (2009). Reaction mechanisms of thiamin diphosphate enzymes: defining states of ionization and tautomerization of the cofactor at individual steps. FEBS Journal, 276(9), 2432-3446. doi: http://doi.org/10.1111/j.1742-4658.2009.06964.x
Noda, Y. & Watanabe, M. (2002). Synthesis of Both Enantiomers of Flavanone and 2-Methylchromanone. Helvetica Chimica Acta, 85(10), 3473-3477. doi: 10.1002/ 1 5 2 2 - 2 6 7 5 ( 2 0 0 2 1 0 ) 8 5 : 1 0 < 3 4 7 3 : AID-HLCA3473>3.0.CO;2-7
Prier, C. K. & Arnold, F. H. (2015). Chemomimetic Biocatalysis: Exploiting the Synthetic Potential of Cofactor-Dependent Enzymes to Create New Catalysts. Journal of the American Chemical Society, 137(44), 13992-14006. doi: http://doi.org/10.1021/%20jacs.5b09348.
Schenk, G., Duggleby, R. G. & Nixon, P. F. (1998). Properties and functions of the thiamin diphosphate dependent enzyme transketolase. The International Journal of Biochemistry & Cell Biology, 30(12), 1297–1318. doi: http://doi.org/10.1016/S1357-2725(98)00095-8
Schneider, G. & Lindqvist, Y. (1993). Enzymatic thiamine catalysis: Mechanistic implications from the three-dimensional structure of transketolase. Bioorganic Chemistry, 21(1), 109–117. doi: http://doi.org/10.1006/bioo.1993.1012
Seebach, D. & Corey, E. J. (1975). Generation and Synthetic Applications of 2-lithio-1,3-dithianes. The Journal of Organic Chemistry, 40(2), 231-237. doi: http://doi.org/10.1021/jo00890a018
Seebach, D. (1979). Methods of Reactivity Umpolung. Angewandte Chemie International Edition in English, 18(4), 239-258. doi: https://doi.org/10.1002/anie.197902393
Shen, B., Makley, D. M. & Johnston, J. N. (2010). ‘Umpolung’ Reactivity in Semiaqueous Amide and Peptide Synthesis. Nature, 465(7301), 1027-1032. doi: https://doi.org/10.1038/nature09125
Smith, A. B., Lin Q., Doughty, V. A., Zhuang, L., McBriar, M. D., Kerns, J. K., Brook, C. S., Murase, N. & Nakayama, K. (2001). The Spongistatins: Architecturally Complex Natural Products—Part Two: Synthesis of the C (29-51) Subunit, Fragment Assembly, and Final to (+)-Spongistatin 2. Angewandte Chemie International Edition, 40(1), 196–199. doi: 10.1002/1521-3773(20010105)40:1<196:aid-anie196>3.0.co;2-t
Tittmann, K., Golbik, R., Uhlemann, K., Khailova, L., Schneider, G., Patel, M., Jordan, F., Chipman, D., Duggleby, R. & Hübner, G. (2003). NMR Analysis of Covalent Intermediates in Thiamin Diphosphate Enzymes. Biochemistry, 42(26), 7885-7891. doi: https://doi.org/10.1021/bi034465o
Tylicki, A., Lotowski, Z., Siemieniuk, M. & Ratkiewicz, A. (2018). Thiamine and selected thiamine antivitamins-biological activity and methods of synthesis. Bioscience Reports, 38(1), 1-23. doi: https://doi.org/10.1042/BSR20171148
Wittig, G., Davis, P. & Koenig, G. (1951). Phenanthrensynthesen über intraionische Isomerisationen. Chemische Berichte, 84(7), 627-632. doi: https://doi.org/10.1002/cber.19510840713.
Wynn, R.M., Daviel, J. R., Mengl, M., & Chuang, D.T. (1996). Structure, function and assembly of mammalian branchedchain α-keto acid dehydrogenase complex. In: Patel M.S., Roche T.E., Harris R.A. (eds) Alpha-Keto Acid Dehydrogenase Complexes. (pp. 101-117). Switzerland: Birkhäuser Basel. doi: https://doi.org/10.1007/978-3-0348-8981-0.
Wynn, R., Kato, M., Machius, M., Chuang, J. L., Li, J., Tomchick, D. R., & Chuang, D. T. (2004). Molecular Mechanism for Regulation of the Human Mitochondrial BranchedChain α-Keto acid Dehydrogenase Complex by Phosphorylation. Structure, 12(12), 2185- 2196. doi: https://doi.org/10.1016/j.str.2004.09.013
Yus, M., Nájera, C. & Foubelo, F. (2003). The role of 1,3-dithianes in natural product synthesis. Tetrahedron, 59(33), 6147-6212. doi: https://doi.org/10.1016/S0040-4020(03)00955-4
Ziegler, F., E., & Schwartz, J.A. (1978). Synthetic Studies on Lignan Lactones: Aryl Dithiane Route to (± )-Podorhizoll and (±)-Isopodophyllotoxone and Approaches to the Stegane Skeleton. Journal of Organic Chemistry, 43(5), 985-991.
